JIA, juvenile idiopathic arthritis.
aPars planitis was characterized as idiopathic uveitis.
There is significant geographic variation with respect to the anatomic distribution, the prevalence of infectious etiologies, and so, the diagnosis of uveitis among children. Ocular toxoplasmosis was seen in only 2% to 10% of all cases among pediatric patients from Western countries (3–5,13,14), while studies from the Middle East and India reported much higher prevalence of this disease among children (12,27). The Indian study found that infectious uveitis was more common in children than in adults and the most common case was pediatric parasitic anterior uveitis (29.6%), followed by endophthalmitis (8%), leptospirosis (5.5%), and toxoplasmosis (4.7%) (25). In the US study, Hispanic ethnicity was associated with a higher prevalence of infectious uveitis, including toxoplasmosis (14). The most common causes of pediatric uveitis based on anatomic location are as follows: anterior uveitis, JIA; posterior uveitis, toxoplasmic retinochoroiditis; and intermediate uveitis, pars planitis (11).
There have been discernable changes in the patterns and anatomic distribution of pediatric uveitis over the last several decades, notably an increase in panuveitis and the decrease in posterior uveitis, presumably reflecting a decrease in toxoplasmosis and toxocariasis in children in developed countries (5). Despite more sophisticated diagnostic techniques, up to 60% of pediatric patients still fall under the category of idiopathic uveitis (1,3,10–15). In our study, the largest number of cases of idiopathic uveitis occurred within the intermediate uveitis group (100%), followed by posterior uveitis (53%), panuveitis (51%), and anterior uveitis (34%) (5). Systemic associations were found in 114 patients (42%), with the most common systemic disease being JIA, accounting for 33% of all cases (5). Table 32.1 illustrates these differences in anatomic distribution, diagnostic associations with systemic disease (JIA), infectious agents (toxoplasmosis), and idiopathic disease and variance with respect to geographic location among 20 pediatric uveitis studies conducted over a 56-year period.
Most pediatric patients at tertiary referral centers have bilateral involvement (1,3–5,12–15) with a mean age at diagnosis between 8 and 10 years of age and a range from 1 to 16 years old (4,5,14). Up to 87% of affected children have a chronic or recurrent disease (5,14). Frequently, there is a delay between the diagnosis of uveitis and presentation to uveitis specialists ranging from <6 months to 13.95 years (5,14,26). Up to 22% of children with JIA-associated uveitis were already legally blind upon their presentation to a tertiary center (26).
The differential diagnosis of pediatric uveitis may be divided into three broad etiologic categories: infectious, noninfectious or endogenous uveitis of autoimmune or idiopathic origin, and masquerade syndromes (Table 32.2). The most common anterior infectious uveitis is from herpes simplex virus (HSV) or varicella– zoster virus (VZV), whereas toxoplasmosis is the most common cause of posterior infectious uveitis. The most common form of noninfectious, endogenous uveitis in childhood is JIA. Kawasaki disease, TINU syndrome, and postviral uveitis are uncommon forms of endogenous uveitis and predominate in the pediatric, compared to the adult, age group. The most important pediatric masquerade syndromes include primary intraocular neoplasms, such as RB, and ocular manifestations of systemic malignancies, such as leukemia.
TABLE 32.2
Types of uveitis in children
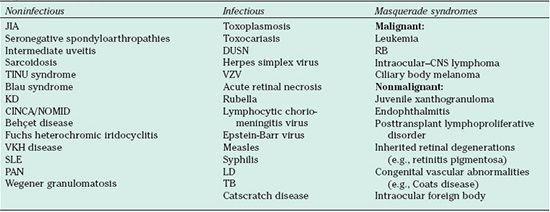
JIA, juvenile idiopathic arthritis; DUSN, diffuse unilateral subacute neuroretinitis; RB, retinoblastoma, TINU, tubulo-interstitial nephritis uveitis; VZV, varicella zoster virus; KD, Kawasaki disease; CNS, central nervous system; CINA/NOMID, chronic infantile neurologic cutaneous and articular/neonatal onset multisystem inflammatory disease ; VKH, Vogt-Koyanagi-Harada disease; SLE, systemic lupus erythematosus; LD, Lyme disease; TB, tuberculosis; PAN, polyarteritis nodosa.
The same anatomical categories of uveitis based on anatomic classification (anterior, intermediate, posterior, panuveitis); grading of anterior chamber cell, flare, and vitreous haze; and descriptors of the onset (sudden or insidious), duration (limited or persistent) and clinical course of uveitis (acute, chronic, recurrent) as defined by the Standardization of Uveitis Nomenclature (SUN) Working Group for adults apply to ocular inflammatory disease in children (27). The extensive differential diagnosis of pediatric anterior uveitis may be somewhat simplified by dividing the disease entities into nongranulomatous and granulomatous categories (Table 32.3). Nongranulomatous entities, such as JIA, herpetic diseases, and human leukocyte antigen (HLA)-B27–associated iridocyclitis, may be distinguished from granulomatous diseases, such as sarcoidosis, inflammatory bowel disease, syphilis, and Lyme disease (LD). Pediatric intermediate uveitis, including pars planitis, is characterized by anterior vitreal inflammation, vitritis and pars plana exudation, and the frequent occurrence of optic nerve head inflammation and cystoid macular edema. Both the infectious and noninfectious entities listed are important considerations in the differential diagnosis (Table 32.3). Using the extended international uveitis study group classification system, pediatric posterior uveitides may be further subdivided between those entities frequently associated with retinal vasculitis and those in which vasculitis is not commonly present (Table 32.3). Entities in which vasculitis is common include toxoplasmosis, sarcoidosis, syphilis, LD, necrotizing herpetic vasculitis, and systemic vasculitic syndromes. Posterior uveitides without a prominent vasculitic component include toxocariasis, tuberculosis (TB), Vogt-Koyanagi-Harada (VKH) syndrome, and presumed ocular histoplasmosis syndrome. Many of the aforementioned disease entities may also produce panuveitis affecting all anatomic regions of the eye (Table 32.3).
TABLE 32.3
Differential diagnosis of pediatric uveitis based on anatomic location
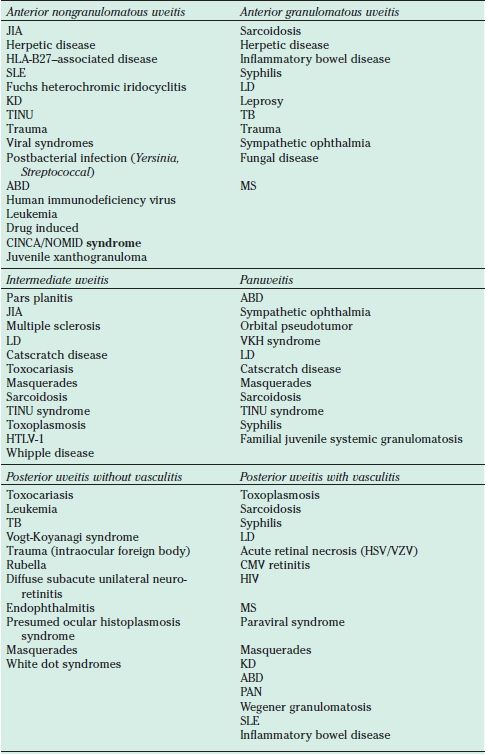
JIA, juvenile idiopathic arthritis-associated iridocyclitis; HLA, human leukocyte antigen; SLE, Systemic lupus erythematosus; LD, Lyme disease; KD, Kawasaki disease; TINU, Tubulointerstititial nephritis and uveitis; TB, tuberculosis; ABD, Amantiades-Behcet disease; MS, Multiple sclerosis; CINCA/NOMID, chronic infantile neurologic cutaneous and articular/neonatal-onset multisystem inflammatory disease syndrome; VKH, Vogt Koyanagi Harada; HTLV-1, human T-cell lymphotrophic virus type 1; CMV, cytomegalovirus; PAN, polyarteritus nodosa; HIV, human immunodeficiency virus; VZV, varicella–zoster virus; HSV, herpes simplex virus.
As a complement to the SUN classification, it is also useful to think of pediatric differential diagnoses in terms of the age of presentation (Table 32.4). It is far more likely for an infant with intraocular inflammation to have a congenital infectious syndrome such as rubella or a malignant masquerade such as RB, while a child of 2 to 10 years of age would be more apt to have an acquired infection such as toxocariasis, Lyme, catscratch disease, or an underlying systemic condition, such as JIA. Adolescents may present with toxoplasmic retinochoroiditis, pars planitis, or even multifocal chorioretinal disease, which are distinctly uncommon in younger age groups. Finally, it must be borne in mind that many of these noninfectious and infectious disease entities may present at any childhood age, including VKH and sympathetic ophthalmia.
TABLE 32.4
Differential diagnosis of intermediate and posterior pediatric uveitis: age at presentation

LCMV, lymphocyctic choriomeningitus virus; RB, retinoblastoma; JIA, Juvenile idiopathic arthritis-associated iridocyclitis; LD, Lyme disease; SSPE, Subacute sclerosing panencephalitis; CINCA/NOMID, Chronic infantile neurologic cutaneous and articular/neonatal-onset multisystem inflammatory disease syndrome; HLA, human leukocyte antigen; CMV, cytomegalovirus; TINU, Tubulointerstititial nephritis and uveitis; TB, tuberculosis; VKH, Vogt Koyanagi Harada; ABD, Amantiades-Behcet disease; HIV, human immunodeficiency virus; VZV, varicella–zoster virus; HSV, herpes simplex virus.
A comprehensive medical history and review of systems, together with a complete ocular examination, employing general anesthesia, as necessary, form the cornerstones to the diagnostic approach in pediatric uveitis. Laboratory tests and ancillary investigations are guided by the above and by the putative differential diagnosis. Laboratory tests are used in most cases to confirm the clinical impression or to exclude diagnostic possibilities, especially infectious entities, rather than serve as a “diagnostic battery.” For example, Rosenbaum and Wernick (28) found that routine screening of all uveitis patients for antinuclear antibodies (ANAs) resulted in approximately 100 false-positive results for every one positive test result in patients with systemic lupus erythematosus (SLE). Therefore, disease-specific testing should be performed in those patients in whom there is a relatively high degree of suspicion for particular diagnoses as suggested by history and examination (Table 32.5).
TABLE 32.5
Workup guidelines for different types of uveitis
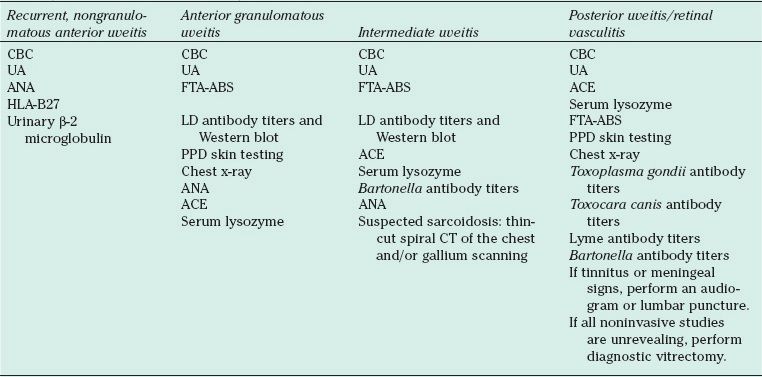
CBC, complete blood count; UA, urinalysis; ANA, anitnuclear antibody; FTA-ABS, fluorescent treponemal antibody absorption; HLAB27, human leukocyte antibody B27; ACE, angiotensin converting enzyme; LD, Lyme Disease; PPD, purified protein derivative; ANA, antinuclear antibody; CT, computed tomography;
COMPLICATIONS OF PEDIATRIC UVEITIS
The literature is replete with reports documenting numerous ocular complications associated with uveitis in children (3–5,11,13,14). It is thought that pediatric population is at greater risk than adults for the development of ocular complications due to the chronicity of inflammation, a lack of symptoms, and longer delays between diagnosis and initiation of appropriate treatment. Complications include band keratopathy, secondary glaucoma, posterior synechiae, cataract formation, cystoid macular edema (CME), vitreous hemorrhage, rubeosis, epiretinal/ neovascular membranes, hypotony, papillitis, papilledema, retinal detachment, and phthisis. The risk and development of amblyopia among young children with uveitis and media opacity is a potential complication, which demands aggressive medical and surgical treatment. The presence of complications at initial examination is an important risk factor for the development of subsequent complications (29).
The most common complications in children with uveitis include posterior synechiae, band keratopathy, cataract, glaucoma, cystoid macular edema, and hypotony (4,5,14,15). Posterior synechiae were found in up to 58% of patients with uveitis, band keratopathy in up to 46%, and cataracts in up to 64%; glaucoma was diagnosed in up to 25% of pediatric patients with uveitis, cystoid macular edema developed in up to 35% of patients, and hypotony in up to 25% (4,5,14,15). Up to 94.6% of children with uveitis evaluated and treated at tertiary centers developed at least one complication (,4,5,14,15). Children with JIA-associated uveitis were found to be at a higher risk of complications than those with other forms of uveitis with the risk of the development of any ocular complication occurring at a rate of 0.33 per eye-year (3,4,5,26,30). Cataract, glaucoma, band keratopathy, posterior synechia, and hypotony occurred at a higher rate among patients with JIA-associated iridocyclitis (5) as compared with other anatomic categories of inflammation. Maculopathy (including cystoid macular edema) and epiretinal and neovascular membranes were more common in intermediate uveitis group (4,5), while papilledema and papillitis occur mostly in patients with posterior uveitis and panuveitis (5).
The presence of anterior chamber flare as measured by laser flare photometry may be relevant to chronic anterior uveitis in children, not merely as a sign of blood–aqueous breakdown but as dynamic sign of disease activity, with a correlation between laser flare values and complications at baseline and over time, as well as outcomes in children with chronic anterior uveitis (31). In a recent study of 114 children with chronic anterior uveitis by Holland et al. (32), baseline and first aqueous flare values were associated with complications and were not a function of disease duration. High baseline and first flare values were associated with an increased risk of visual loss to a level of <20/50 and in the development of new vision-threatening complications independent of the presence of anterior chamber cells. Anterior chamber flare may emerge as an important marker of projected disease severity, indicative of long-term risk, changing little over time. Anterior chamber cells, on the other hand, may be more indicative of present disease status and reflective of short-term changes with greater response to treatment.
It is not surprising that vision loss has been reported to be more common in pediatric uveitis than that in adults given the high rate of complications and the chronicity of disease seen in many childhood entities. Series from the 1960s reported that 30% of children with chronic uveitis became blind (16). A more recent review of literature reported that one-fourth to one-third of all children with uveitis had severe vision loss (11). In a study from Bascom Palmer Eye Institute, the most frequent causes of vision loss to 20/200 or worse were cataract, vitreous opacification, cystoid macular edema, and a macular choroidal neovascular membrane or scar (4). In spite of the fact that some vision loss was later corrected by surgery or medical therapy, the cumulative proportion of vision loss of 20/200 or worse was 9.7% at 5 years of age and 22.8% at 8 years of age. The median age at time of vision loss to 20/200 or worse was 13 years in this study (4).
In the series of patients with pediatric uveitis from the Netherlands, 17% had a visual acuity (VA) of 20/200 in at least one eye, while 2% had this level of acuity bilaterally (13). In an older study, we found 20/300 vision in 63 of 137 eyes (46.0%) followed for at least 6 months (33). More recently, in a study on JIA-associated uveitis, we found that 22% of eyes were legally blind at presentation (26). At the final follow-up visit, 18% of eyes (25 patients) had vision of 20/200 or worse. Of the 25 patients with poor vision at the time of data analysis, 9 were seen only twice or less. The mean time between the onset of uveitis and first examination by us (cerebrospinal fluid [CSF]) was 5.01 years, underscoring the paramount importance of early referral and intervention to significantly improve these devastating statistics.
A poor visual prognosis has been traditionally associated with increased severity of JIA-associated uveitis at initial ocular examination including the presence of baseline complications, longer duration of uveitis, younger age at presentation, and longer time before referral to a uveitis specialist (26,34). There are, however, several published reports citing better visual outcomes of patients with JIA-associated uveitis (27,34–36). A British study found that only 6% of eyes of 163 patients ended with blindness (37). None of 63 patients in a Finnish study went blind from JIA-associated uveitis (37). Oren et al. (34) reported on 10 children, all of whom had final VA of 20/30 or better. An Italian study of JIA found that 64.5% of their patients maintained VA of 20/33 or better at final follow-up (37). Furthermore, this disparity in the literature with respect to the visual outcome in patients with JIA-associated iridocyclitis is highlighted by the more favorable outcomes reported among population-based studies, and surveys of JIA-associated iridocyclitis in a large pediatric hospital with a visual acuity of 20/40 or better have been reported in 87% of patients (39,40).
This is in stark contrast to the poorer outcomes reported from tertiary care ophthalmic centers where the visual prognosis remains guarded for children with JIA-associated uveitis (37). Indeed, an estimated frequency of visual loss over 3 years of 30% to a level of 20/50 and 24% to a level of 20/200 was reported among patients with JIA-associated iridocyclitis followed at the Wilmer Eye Institute (30). In our more recent study on pediatric uveitis, the changes in VA in 469 eyes were documented on each visit (5). Visual acuity at the time of the diagnosis of uveitis was available for all except two children. Visual acuity of 20/40 or better was found in 71% of all eyes at the time of diagnosis, 20/50 to 20/200 in 20% of the eyes, and 20/300 to light perception (LP) with correct light projection in 8%. There was no LP in two eyes at the time of diagnosis. At the final follow-up with us (CSF), VA was 20/40 or better in 77% of eyes, 20/50 to 20/200 in 17%, and 20/300 to LP in 5% of eyes. Seven eyes (1.5%) ended with no LP (5). We analyzed the relationship between visual outcomes and the time between diagnosis of uveitis and referral to uveitis specialist. The longer the time before patients were seen by the uveitis expert, the worse were the visual outcomes (5). There was a tendency to refer patients diagnosed with posterior uveitis and panuveitis to the tertiary center sooner (means, 11 and 7 months, respectively) than patients diagnosed with intermediate uveitis (19 months) and anterior uveitis (33 months) (5). Several studies conclude that posterior uveitis is a risk factor for poor visual outcome (3,4,14).
It is our view that visual prognosis for pediatric uveitis is potentially modifiable by early diagnosis and referral to a uveitis specialist with appropriately aggressive anti-inflammatory treatment, antimicrobial therapy in cases of infectious entities, and the early introduction and sustained use of steroid-sparing IMT for chronic noninfectious uveitis. The use of new diagnostic modalities such as laser flare photometry together with a reassessment of the current screening guidelines may serve to identify those eyes that are at greatest risk for development of complications and visual loss and might benefit most from an aggressive therapeutic approach.
SPECIFIC UVEITIC DISORDERS IN CHILDREN
Noninfectious Uveitides
Juvenile Idiopathic Arthritis-Associated Iridocyclitis
JIA, a multisystemic autoimmune disease, is the most common form of arthritis of childhood affecting approximately 70,000 children in the United States and is the most common identifiable systemic association with iridocyclitis, with an average cumulative incidence of approximately 8.3%, representing 2,500 to 5,000 cases and up to 38% of those seen in a tertiary care setting (39,41). Its pathogenesis appears to be multifactorial, as in most autoimmune diseases, with genetic predisposition having been identified and the onset possibly triggered by microbial contact. The peak age of onset of JIA is between ages 6 months and 4 years with three broad modes of onset: systemic, oligoarticular, and polyarticular. The International League of Associations for Rheumatology (ILAR) classifies JIA into the following categories with the attendant associated risk of uveitis: systemic (rare); oligoarticular, persistent, or extended (10% to 30%); rheumatoid factor (RF)–negative polyarthritis (5% to 10%); RF-positive polyarthritis (rare, scleritis); psoriatic (10% to 20%); and enthesitis related (10% to 15%, HLA-B27, acute, recurrent, uniocular) (35).
The uveitis associated with JIA is typically silent, with the patient being completely asymptomatic and unaware of the presence of intraocular inflammation. This is an extraordinarily curious aspect of the disorder, since the vast majority of other individuals who develop uveitis experience pain, ocular redness, light sensitivity, and/or diminished vision. While typically the onset of arthritis precedes that of intraocular inflammation, the converse may be true with as many as 10% having chronic intraocular inflammation for prolonged periods of time prior to entering into the health care system and resulting in delay in diagnosis, treatment, and poorer outcome. Furthermore, the systemic and ocular inflammatory activity may not be synchronous, and their management should be considered independently. In addition to JIA subtype (oligoarticular disease), risks of the development of uveitis include ANA positivity, younger age at onset of arthritis (2 to 5 years), female sex, and HLA-DR5 positivity, while predictors of poor visual prognosis were associated with increased severity of ocular disease at presentation and include active inflammation, presence of posterior synechiae, abnormal IOP, uveitis onset before or concurrent with diagnosis of arthritis, shorter interval from onset of arthritis to uveitis, prolonged referral to a uveitis specialist, and male gender (5,26,27,30,42–46).
Ocular structural complications associated with visual loss include cataract, band keratopathy in the visual axis, posterior synechiae, hypotony, glaucoma, and macular edema, optic nerve edema, vitreous haze, and epiretinal membrane with an incidence of any complication occurring at 33% per eye per year (30). Ocular complications at presentation are common, being observed in up to 45% of children with JIA in a large population-based study (47) and in 67% of 75 patients with JIA followed over a 20-year period at the Wilmer Eye Institute (48). In the latter study, complications at presentation were associated with active inflammation, ANA positivity, and a shorter duration between the diagnosis of arthritis and uveitis. Poor vision at presentation due to cataract, central band-shaped keratopathy, or glaucoma to a level of 20/50 was seen in 36% of patients and 20/200 in 24%. The presence of active anterior segment inflammation and a history of previous ocular surgery before presentation were significantly associated with both levels of visual loss, while the additional finding of posterior synechiae at presentation was associated with 20/200 (legal blindness) (48).
Regrettably, even today, with ophthalmologists and rheumatologists alike following the recommendations of the American Academy of Pediatrics (49) for biomicroscopic screening of children with JIA, the proportion of severe disease presentation appears to be unchanged with as many as 25% developing significant visual impairment and, in some regions, as many as 12% becoming blind in at least one eye (50). This fact is underscored by the publication by Ozdal et al. (51), in which final visual acuity in their JIA population was imperfect in 40%, quite poor in 20%, and completely lost in 10% of eyes. The authors conclude that 70% of the eyes of the patients that they observed were either visually handicapped or totally blind, and the authors emphasize that some of the publications purporting a more favorable ocular prognosis in children with JIA-associated uveitis may suffer from case selection bias and by inadequate longitudinal follow-up observations. Although Kanski (52), Chylack (53), and Wolf et al. (54) reported poor outcomes in their JIA populations 25 to 30 years ago, Sherry et al. (55) reported that the prevalence of uveitis had been steadily decreasing as a consequence of better JIA management by rheumatologists and that severe visual loss had dropped from 21% in 1975 to none at the time of the 1991 publication. Chalom et al. (56) emphasized that the outcome of their pediatric population of JIA who developed uveitis was excellent 95% of the time in a 1997 publication. Kotaniemi et al. (38) reported ocular complications in 24% of their patients, but an overall good outcome visually in 97% in a publication on this matter in 2001. However, Cabral et al. (57) emphasized that the occurrence of ocular complications associated with JIA-associated uveitis was common (32%) and vision impairment was not rare (15%).
Ozdal et al. studied 18 patients with JIA-associated uveitis referred to the Royal Victoria Hospital, McGill University Health Center in Montreal, Canada. All patients had ocular complications at the time of first evaluation at the clinic at McGill, with visual acuity <20/150 in 30% of the patients and three eyes in phthisis (51). These authors emphasize that all of the patients were adults and that, therefore, the uveitis specialists at the McGill clinic were seeing the consequences of the follow-up of the patients with very long-standing uveitis, with a mean duration of the disease in their patient population of 24.9 years and with the mean duration of the uveitis of 20.5 years. The authors conclude that the long-term prognosis of ocular involvement in patients with JIA-associated uveitis is still poor, with final visual acuity of 20/50 or worse in 70% of the eyes. They emphasize that outcomes would be improved only with earlier disease detection and increased vigor of therapy by physicians skilled in the use of immunomodulatory agents. We support this opinion as far too many children with JIA-associated uveitis lose vision to the point of being permanently visually handicapped, as a consequence of delayed diagnosis and as a consequence of inadequate treatment. A study from the Bascom Palmer Eye Institute, University of Miami, reported 34 JIA-associated uveitis patients in a group of 149 pediatric uveitis cases followed for up to 10 years with band keratopathy, glaucoma, cataract, macular edema, and hypotony in many, finding that the cumulative proportion of patients with complications continued to rise through time, with substantial numbers of eyes and patients with visual acuity of 20/200 or less in the better eye (4). As previously mentioned, the disparity in the literature with respect to visual outcome in patients with JIA-associated iridocyclitis, with a more favorable outcomes reported among population-based studies (39) and surveys of JIA-associated iridocyclitis referred to pediatric hospitals (40) as opposed to poorer outcomes in our experience (26) and those reported from other tertiary care ophthalmic centers (4,21,51), may be more the reflection of sampling bias and lack of long-term follow-up, obfuscating the harsh reality of significant visual debility often associated with this disease.
Our therapeutic approach to JIA-associated iridocyclitis is not unlike our approach to other forms of noninfectious uveitis and includes the elimination of all active inflammation with limited tolerance for steroids and the early introduction of steroid-sparing IMT. Initial treatment with topical steroids commenced with regional corticosteroid injections (generally requiring anesthesia) being added to the program if the uveitis is not abolished through the frequent application of topical steroids. Brief (no >3 months) systemic corticosteroid therapy may also be employed. We use a so-called stepladder algorithmic paradigm in our care of patients with JIA-associated uveitis, with steroid therapy representing the first step on the stepladder and chronic oral nonsteroidal anti-inflammatory drug (NSAID) therapy representing step 2, employed in instances in which the uveitis recurs with every attempt at tapering the topical steroids. A variety of oral NSAIDs may be used, but the two with the longest track record in the hands of pediatric rheumatologists are naproxen and tolmetin. The usual caveats pertain, of course, with respect to NSAID use, and the therapy should be coordinated with the patient’s pediatrician and/or pediatric rheumatologist. Some patients with JIA-associated uveitis have recurrences of the uveitis despite the chronic use of NSAIDs. These patients must be advanced to the third step on the stepladder: IMT.
A variety of immunomodulatory medications have been used in this context; however, the antimetabolite methotrexate has, by far, the longest safety and efficacy record and is the agent of first choice in the treatment of pediatric uveitis in general and in JIA-associated iridocyclitis specifically. We typically commence therapy at a dose of 0.15 mg/kg body weight once weekly by mouth and ask that the child also take 1 mg of folic acid per day, a strategy that has been clearly shown to reduce the likelihood of hepatotoxicity in patients receiving methotrexate (58). We advance the dose of methotrexate, as needed and tolerated, to achieve the goal of complete freedom of all recurrences of all inflammation at all times off all steroids, making the dose changes approximately every 6 to 8 weeks. Curiously, the uveitis associated with JIA often requires much higher doses of methotrexate to induce a durable remission of the uveitis than does the arthritis associated with JIA. Fortuitously, children tolerate such doses of this medication quite well. The bioavailability of the medication is variable after oral administration; therefore, we switch patients to subcutaneous administration in the event that the uveitis persists despite multiple increases in the orally administered dose. We make this switch whenever the dose escalation rises above 17.5 mg.
Potential side effects of methotrexate use include hepatotoxicity, bone marrow suppression, interstitial pneumonitis, mucositis, nausea, fatigue, and alopecia. Most of these potential adverse effects are medically trivial, but hepatotoxicity, interstitial pneumonitis, and bone marrow suppression are not. It is for this reason that it is mandatory that the patient be regularly monitored by an individual who is, by virtue of training and experience, truly expert in the prescribing and monitoring of IMT, in general, and methotrexate, in particular. We monitor our patients every 6 weeks, both by personal observation and query, and by hematologic assay (alanine aminotransferase [ALT] and aspartate aminotransferase [AST] liver enzymes, blood urea nitrogen [BUN] and creatinine, and complete blood count [CBC]). Suppression of the bone marrow, in our experience, is extraordinarily rare. Rising liver enzymes is an indication of hepatotoxicity, and while rheumatologists, in general, tolerate a 30% rise above the upper limits of normal before reducing the methotrexate dose by 50%, our tolerance for rising liver enzymes is less, and we typically back off on the dose of the medication if the patient has elevated enzymes on two occasions separated by 2 weeks. Interstitial pneumonitis is characterized by diminished exercise tolerance and by development of cough. It, like the chemical hepatitis, is reversible if detected early in the course of its development, with cessation of use of the drug that caused the onset of the problem.
We currently treat our JIA-associated uveitis patients who are eventually advanced to methotrexate therapy for a minimum of 2 years, after the uveitis has been in remission, off all steroids. Inflammatory control had been observed in 60 to 82% of patients with a steroid-sparing effect in >50% of patients (37,59,60). A recent study confirmed our clinical impressions and practice in that inflammatory relapse was observed in 69% of patients within 1 year of withdrawal of medication. Both a longer period of inactivity prior to withdrawal and longer treatment periods with MTX drug for up to 3 years are associated with a reduction of the chance of relapse after drug discontinuation (61). This means, of course, that 20% to 40% of patients do not succeed with methotrexate as monotherapy. For them, alternative agents are explored, until the agent or combination of agents is found that accomplishes the aforementioned goals for them. This trial-and-error exercise can be extremely frustrating for patient, family, and doctor alike. However, to give up on the quest for disease remission is, in our view, an enormous mistake. One must stay in the hunt, never quitting on the patient, and prevailing over the problem, in the end.
In those patients not responding to methotrexate, alternative antimetabolites such as azathioprine (3 mg/ kg/day) or mycophenolate mofetil (1,200 mg/m2/day) may be substituted, cyclosporine (up to 5 mg/kg/day) may be used as monotherapy or in combination with antimetabolites, or intravenous immunoglobulin (2 g/ kg/ cycle, once monthly) has been employed. While we have used cytotoxic agents such as chlorambucil (0.1 mg/kg/ day) and intravenous cyclophosphamide (1 g/m2 once monthly) in the treatment of recalcitrant childhood uveitis, they are employed infrequently due to very real concerns regarding their serious long-term side effects.
The introduction of biologic response modifiers, particularly the tumor necrosis factor (TNF) inhibitors infliximab and adalimumab, has added a new dimension to our treatment armamentarium, and these biologic response modifiers are the agents to which children with methotrexate-resistant JIA-associated iridocyclitis are advanced. Etanercept, while effective in treating the articular manifestations of JIA, has mixed results with respect to treatment of iridocyclitis, has been associated with de novo inflammation, and has been shown to be ineffective both in preventing relapses and in the treatment of JIA-associated iridocyclitis in two small, randomized controlled studies (62,63). Moreover, etanercept has been shown to be differentially less effective in the treatment of intraocular inflammation and other TNF inhibitors (64). Therefore, it is currently recommended that patients with new-onset or incompletely controlled uveitis who are on etanercept be switched to either infliximab or adalimumab (40).
infliximab (5 mg/kg monthly after induction, up to 10 or even 20 mg/kg if needed and tolerated) has demonstrated consistent anti-inflammatory activity at higher doses and shorter infusion intervals than that used for the treatment of rheumatoid arthritis, allowing a reduction in the steroid requirement for both topical and systemic steroids and permitting systemic immunomodulatory taper (65–68). As this monoclonal antibody is a mouse–human chimera, it is recommended that patients be maintained on low-dose antimetabolite therapy for the prevention of human antichimeric antibodies. Unfortunately, infliximab does not consistently produce disease remission with its efficacy waning over time (69).
Adalimumab (40 mg q 14 days up to q 7 days if needed) is a potentially effective treatment option after methotrexate failure or may be considered as maintenance therapy following induction with infliximab (68,70–72). The advantage to this medication is that adalimumab is fully humanized, and so, less immunogenic, and may be administered subcutaneously rather than intravenously. The optimal dosage and treatment protocols are not defined and must be individualized for each patient.
While the formation of anti-DNA antibodies may rarely produce a drug-induced lupus syndrome with infliximab, more significant concerns surrounding long-term use of the agent include an increased risk of infection including TB and histoplasmosis, demyelination, retrobulbar optic neuritis, heart failure, and a potentially increased risk of lymphoma in children and young adults (73–76,559). This has led the FDA to issue a “black box” warning for these medications with respect to malignancy risk in this setting. Caution must be exercised in patients with intermediate uveitis or pars planitis given the associated risk for the development of demyelinating disease. TNF inhibitors are contraindicated in patients with symptoms or magnetic resonance imaging (MRI) findings consistent with multiple sclerosis (MS).
Most recently, the anti–B cell monoclonal antibody rituximab has been shown to control uveitis in 7 of 10 patients with JIA-associated iridocyclitis refractory to IMT and other anti-TNF agents (77). Similarly abatacept, a selective T-cell co-stimulation molecule, was found to achieve remission in six of seven patients with refractory JIA-associated iridocyclitis with a reduction in the frequency of uveitis flares following treatment (78).
The complications of JIA-associated iridocyclitis may require surgical management. Cataract and glaucoma, in particular, occur in many patients with this disorder, and the glaucoma that develops is frequently resistant to adequate control medically. Approximately 40% of the patients with JIA-associated uveitis referred to us already have glaucoma at the time of first referral. They generally also have cataract. Therefore, the decision-making becomes complex. Should the cataract come out? Should the patient have a lens implant at the time of cataract removal? Should a glaucoma procedure be performed simultaneously? If the patient is judged to be a poor risk for the presence of an intraocular foreign body such as an implant, should contact lens wear or aphakic spectacles or both be planned? Should a mitomycin-C trabeculectomy or a valve tube shunt be employed for the glaucoma surgery? Should the surgery be performed soon or deferred until better control of the uveitis can be accomplished? This latter question is especially challenging in the child who still has the potential for avoidance of amblyopia. However, of course, operating on the inflamed eye, particularly performing cataract surgery, is notorious for poor outcomes. Therefore, the urgency of prevailing over the active uveitis is even more palpable.
Historically, the experience of most uveitis specialists has been that children with JIA-associated iridocyclitis are poor candidates for the incorporation of a lens implant (79–81). The formation of inflammatory membranes behind or around the implant, with membrane contraction and progressive hypotony, has resulted in many such eyes being destroyed by phthisis. It is also believed that the best approach to this problem is cataract removal and the performance of a total vitrectomy, with aphakic soft contact lens fitting subsequently (82–84). It follows, therefore, that in such a patient with coexistent glaucoma, a trabeculectomy would be a poor choice for the glaucoma management, since the risk of endophthalmitis in a contact lens wearer with a filtering bleb is unacceptable. Therefore, for coexistent glaucoma, we would favor the use of an Ahmed valve tube shunt (85,86), and this is generally placed through the pars plana. We think that in well-selected cases, with adequate control of perioperative inflammation and proper follow-up, an intraocular lens (IOL) can be well tolerated. Advances in surgical techniques and devices in cataract surgery have popularized primary placement of an IOL in the pediatric population. A multicenter analysis by Nemet et al. (87) found no significant difference in postoperative course or complications when comparing non–JIA-associated uveitis patients and JIA. Lam et al. (88) reported favorable visual outcomes after cataract surgery with primary IOL implantation in children with JIA-associated uveitis. We described improved postoperative acuity in 92% of pediatric patients and found that placement of IOL did not significantly raise the level of postoperative inflammation (89). However, prudence absolutely dictates that one be very thoughtful on this matter, counseling parents carefully, preparing them for the possibility for the need for additional surgery for IOL explanation in the event that their child demonstrated that the presence of the IOL was clearly producing adverse consequences that threaten vision over the long term.
Seronegative Spondyloarthropathies
The seronegative spondyloarthropathies include ankylosing spondylitis (AS), reactive arthritis, psoriatic arthritis, and inflammatory bowel disease that share many clinical features, including a predisposition to sacroiliitis and to uveitis and the HLA-B27 gene.
Ankylosing Spondylitis: Pediatric ankylosing spondylitis (AS is a well-characterized syndrome. As with the adult form of the disorder, HLA-B27–positive males predominate in the pediatric population. The arthralgia begins insidiously, generally with morning low back stiffness, with (eventually) the evolution of dull low back pain, which improves throughout the course of the day. Direct pressure over the sacroiliac joints often elicits pain, and the patient is found to have limited lower-spine flexibility, with inability to touch his or her toes without bending the knees. Peripheral arthritis may also occur (usually knees, shoulders, or hips), and as with JIA, the temporomandibular joint (TMJ) may be involved. Indeed, this may be the single peripheral joint involved, and one must avoid reflexively ascribing TMJ symptoms to “TMJ syndrome” rather than to true arthritis. Enthesopathy (inflammation of tendons, particularly at their points of insertion into bone) is highly typical of AS, with exquisite tenderness in the areas of tendon inflammation.
Juvenile-onset spondyloarthropathies, by definition, occur in children under the age of 16. AS typically begins after the age of 10 years. By definition (“seronegative”), the patients are RF negative. The anterior uveitis associated with AS (indeed with all of the seronegative spondyloarthropathies) is generally acute, unilateral, and recurrent, but eventually becomes bilateral, alternating. As many as 15% of children with juvenile AS may develop chronic iridocyclitis (90). HLA-B27 positivity is not required for the diagnosis; 5% to 10% of patients with AS are HLA-B27 negative. Unlike the uveitis associated with JIA, the uveitis associated with the seronegative spondyloarthropathies is often symptomatic, with patients complaining of red eye, ocular discomfort, and photophobia. Therapy should include aggressive topical corticosteroids (every 30 to 60 minutes while awake) along with cycloplegia; however, in the presence of severe inflammation (fibrin) or when topical therapy does not result in a significant reduction in the intensity of anterior chamber reaction, a short burst of systemic corticosteroid (1.5 mg/kg/day), with a rapid taper (dosage reduction every 5 to 7 days), is appropriate. More recalcitrant cases may require regional corticosteroid injections, and this generally requires brief general anesthesia in children.
HLA-B27–associated uveitis tends to be recurrent, and our experience suggests that oral NSAIDs are particularly useful in long-term management of patients with nongranulomatous, acute idiopathic, or HLA-B27–associated recurrent anterior uveitis, allowing a reduction in the amount of corticosteroid required to achieve inflammatory quiescence and enabling patients, in many cases, to maintain a steady course without inflammatory exacerbations once steroids have been discontinued (91). specifically, we reported the efficacy of oral NSAIDs in the prevention of uveitis recurrences among 59 patients with recurrent nongranulomatous, idiopathic, or HLA-B27–associated acute anterior uveitis (AAU) (92). The average duration of NSAID therapy was 21.2 ± 5.7 months. The average number of relapses of all patients declined from 2.84 to 0.53 per person-year follow-up prior to and following systemic NSAID therapy respectively. The relapse rate before and after treatment significantly attenuated in both the HLA-B27–positive (n = 21) and HLA-B27–negative (n = 38) subgroups, reducing morbid attacks and the cumulative exposure to corticosteroids. The NSAIDs used in this study were celecoxib, a cyclooxygenase-2 (COX-2) inhibitor, and diflunisal, nonselective COX inhibitor. While the therapeutic effect obtained by each was similar, the side effect profile of celecoxib and diflunisal is quite different, with celecoxib being much better tolerated. For both JIA- and AS-associated uveitis, one should be guided by the child’s rheumatologist as to the choice and dosing of the NSAID.
Reactive Arthritis (Reiter): Reactive arthritis (previously designated as “Reiter syndrome”) was originally defined as the triad of arthritis, conjunctivitis, and noninfectious urethritis, but uveitis may substitute for or be present along with conjunctivitis in patients with reactive arthritis. Reactive arthritis may occur in children. Although it is not extremely uncommon in adults, it certainly is rare in children. Approximately 75% of adults who develop reactive arthritis are HLA-B27 positive, while 90% of children with it are HLA-B27 positive. About 2% of pediatric patients with reactive arthritis develop uveitis. The uveitis observed with this syndrome is identical to that seen with AS as well as with HLA-B27– associated uveitis in the absence of spondyloarthropathy. It is said that approximately 12% of patients with reactive arthritis will develop uveitis with the initial attack being typically unilateral and acute. As with AS, the uveitis associated with reactive arthritis is generally nongranulomatous and recurrent, and hypopyon may occur. Therapy is similar to that described above for AS-associated intraocular inflammation.
Psoriasis: Children may develop uveitis in association with psoriasis, with or without the development of arthritis. Girls are slightly more likely than boys to develop psoriasis (3:2) with a mean age of onset of approximately 9 years. The uveitis associated with psoriasis without arthritis is slightly different from that of uveitis associated with psoriatic arthritis or HLA-B27– associated uveitis (93). The mean age of onset is older, and it tends to be bilateral and of longer duration and require oral treatment with NSAIDs. Retinal vasculitis, macular edema, and papillitis are more frequently seen.
The skin lesions of psoriasis typically precede the development of either joint or ocular inflammation; however, we have made the diagnosis of psoriasis in numerous instances while examining the skin of patients during their initial evaluation of uveitis with subsequent dermatologic referral. Small hidden patches of itchy, scaly dermatitis may come and go, in the axilla, on the umbilicus, on the genitalia, on the back, or on the scalp. The prevalence of uveitis in patients with psoriasis is unknown. Uveitis may occur in up to 20% of pediatric patients with psoriatic arthritis (94,95). As with AS and reactive arthritis, psoriasis-associated uveitis presents with acute, recurrent, nongranulomatous, unilateral inflammation, but with time, nonsimultaneous bilateral involvement may occur. It is more severe and more recalcitrant in patients who are HLA-B27 positive. Patients who develop uveitis associated with psoriatic arthritis are also more commonly ANA positive (80%), with oligoarticular arthritis presenting as long as 10 years before the onset of the dermatologic manifestations of psoriasis; hence, the diagnosis of JIA has often been (erroneously) made earlier in these children.
Therapy is with aggressive topical corticosteroid and cycloplegic and chronic oral NSAID therapy may also be helpful in reducing the likelihood of recurrent inflammation. Treatment-resistant cases respond well to low-dose once weekly methotrexate, daily cyclosporine, and possibly to TNF-α antagonist therapy.
Inflammatory Bowel Disease (Enteropathic) Arthritis: Enteropathic arthritis–associated uveitis may occur in patients with either Crohn disease or with ulcerative colitis. These disorders are associated with peripheral arthritis (20% in patients with Crohn disease and 10% in patients with ulcerative colitis) and with sacroiliitis (10% in patients with either Crohn disease or ulcerative colitis). It is also associated with the HLA-B27 haplotype, but uveitis may occur in patients without enteropathic arthritis (i.e., simply with inflammatory bowel disease) and in patients who have inflammatory bowel disease but who are HLA-B27 negative. Uveitis occurs in 10% of patients with inflammatory bowel disease and is more common in patients with Crohn disease than with ulcerative colitis (94). Episcleritis may be seen in association with anterior uveitis. In most cases, the clinical presentation is similar to that of patients with the other seronegative spondyloarthropathy-associated uveitides as is its treatment. While much less frequent, posterior uveitis may also occur, characterized by granulomatous panuveitis with choroidal infiltrates, retinal vasculitis, serous retinal detachment, retrobulbar neuritis, and papillitis (96–98). Surgical excision of the inflamed bowel may be beneficial in management of the extraintestinal symptoms, including peripheral arthritis and uveitis. Immunomodulatory medication (especially with 6-mercaptopurine) and anticytokine therapy (especially with anti-TNF-α agents) may be critical elements in the eventual successful control of the patient’s intestinal and extraintestinal problems.
Cervical Spondylitis: Cervical spondylitis may occur in young girls, resulting in cervical apophyseal joint fusion and symmetric destructive polyarthritis involving small joints in the hands and wrists. Some of these girls are HLA-B27 positive; others are HLA-B27 negative; most are rheumatoid factor (RF) and anti-nuclear antibody (ANA) negative (65%). Those girls who are HLA-B27 positive and ANA positive, however, develop chronic iridocyclitis quite indistinguishable from that of patients with JIA. Those girls who are HLA-B27 positive but ANA negative typically develop recurrent acute uveitis indistinguishable from that of patients with AS or other “typical” seronegative spondyloarthropathies.
Intermediate Uveitis
Intermediate uveitis is estimated to represent one-fifth of all cases of pediatric uveitis (99). There appears to be no specific predilection for race or gender. The prevalence of pars planitis peaks between childhood and the fourth decade, but older people can also be affected. Seventy to ninety percent of cases are bilateral. About a third of patients with unilateral involvement at the time of presentation develop bilateral involvement. No inheritance pattern has been defined, but isolated reports of familial cases support the idea that this disease may have genetic predispositions (100–109).
Patients with pars planitis generally present with complaints of floaters and blurred vision, typically with a “quiet” and white eye. In some children, intermediate uveitis is found accidentally on routine screening. Pain, redness, tearing, and photophobia are signs and symptoms of anterior segment inflammation, which are seen more commonly in children with pars planitis than in adults. Moderate to severe cells and flare, keratic precipitates, band keratopathy, and even posterior synechiae may be found. Children presenting with idiopathic pars planitis have a worse visual acuity both at initial diagnosis and at follow-up than do adults (110). Posterior subcapsular cataract formation is frequently found at presentation.
Vitreous cells are the most important finding in patients with intermediate uveitis and are the sine qua non for making the diagnosis. Cellular exudates in the vitreous and on the pars plana are seen on depressed peripheral examination of retina in patients with active pars planitis, while old cellular debris, crenated residua of prior vitreal exudates, and a collagen band or “snowbank” are generally present in the patient whose pars planitis is inactive. The extent of “snowbank” may vary from a few clock hours, mostly inferiorly, to 360 degrees of the retinal periphery. The collagen “snowbank” may be acellular or contain fibrous astrocytes. It consists of tenascin and collagen types I, II, and III and reflects the consequences of inflammation involving peripheral retina and the vitreous base (111). While presence of snowbanks or snowballs is not required to make the diagnosis of intermediate uveitis, they are prominent clinical features of pars planitis and serve to delineate this entity as a subset of intermediate uveitis. Presence of snowbanks is associated with worse visual outcome (112,113).
Other signs of the disease include peripheral retinal vasculitis with sheathing of both venules and arterioles. Vasculitis may lead to occlusion and neovascularization of peripheral retina and optic nerve. Vasculitis was found in 10% to 32% of cases (114–116).
Autoimmune endotheliopathy is a rare finding in the anterior segment associated with intermediate uveitis. Peripheral corneal edema evolves, with keratic precipitates lying in a linear fashion, marking the border of edematous cornea from normal cornea. This transplant rejection–like appearance signifies an autoimmune etiology of intermediate uveitis (117–119).
Children are generally at higher risk for development of complications from pars planitis. The higher prevalence of complications in children is explained by a more asymptomatic course of intermediate uveitis compared with that in adults, later referral, and a greater degree of irreversible damage on initial presentation. Complications of pars planitis in children include cataract, macular edema, secondary glaucoma, band keratopathy, retinal detachment, optic neuropathy, neovascularization of the peripheral retina or optic nerve, vitreous hemorrhage, and formation of inflammatory membranes. Amblyopia may develop as a result of prolonged inflammation and the development of these complications.
Cataract develops in about 50% of patients with intermediate uveitis (120). It is associated with both uncontrolled inflammation and with use of corticosteroids. Posterior subcapsular cataract is the most common observed type, highlighting the iatrogenic contribution of corticosteroids to the development of this complication. Cataract formation is less common in patients who were treated with steroid-sparing immunosuppressive medications early in the course of disease (112,120).
Cystoid macular edema and maculopathy are the most common causes of severe visual impairment in patients with intermediate uveitis with an incidence ranging from 12% to 50% (4,5,14). Left untreated, cystoid macular edema leads to permanent loss of vision through evolution of cystic macular edema, macular pucker from epiretinal membrane, and macular hole formation.
Secondary glaucoma occurs predominantly as a result of topical corticosteroid therapy. In most studies, ocular hypertension has not been reported in association with intermediate uveitis (4,121).
In our series, however, 24% of children with pars planitis developed elevation in intraocular pressure (5).
Band keratopathy is considered a hallmark of pediatric uveitis and may be severe, especially in younger children. As it occurs most commonly in the setting of chronic anterior segment inflammation, children with intermediate uveitis develop this complication more often than do adults.
Retinal detachment is a complication seen with considerable frequency in patients with intermediate uveitis. Exudative retinal detachments occur in 5% to 17% of patients with intermediate uveitis (114,120,122,123). Such high prevalence is uncommonly seen in other uveitic entities except for the VKH syndrome. In addition, patients with pars planitis may present with bullous retinoschisis. It occurs mostly in the inferior periphery. Both exudative retinal detachments and retinoschisis may be related to a Coats-like vascular response secondary to chronic inflammation (124). Vitreoretinal traction is reported in 3% to 22% of patients with intermediate uveitis, leading to retinal tears and combined rhegmatogenous–tractional retinal detachments (125). Rhegmatogenous retinal detachment due to uveitis in children has been reported in up to 15% of cases (126).
Optic nerve involvement in intermediate uveitis is observed much more frequently in children than in adults (99,105,106,122,123,127). Disc edema is present in 3% to 50% of eyes (106,122,123,128). Long-standing edema may lead to optic nerve atrophy, while extensive retinal ischemia may result in optic disc neovascularization. Optic neuritis in pars planitis patients occurs in association with MS in up to 38.5% of cases (129,130). Even though MS is exceptionally rare in children, one might consider a systemic workup if a patient is a female, has a bilateral intermediate uveitis, and is HLA-DR15 positive.
Peripheral retinal neovascularization is commonly reported in patients with intermediate uveitis and may become vision threatening with the development of recurrent vitreous hemorrhages. In many instances, vitreous hemorrhages will clear, but nonclearing hemorrhages require surgical intervention (116). Children with pars planitis are more likely than adults to experience vitreous hemorrhage. Peripheral retinal neovascularization may lead to formation of vascular cyclitic membranes. Although epiretinal membranes in pars planitis are rarely considered visually significant, their presence is grossly underestimated. The ability to detect epiretinal membranes before surgery was nearly a half of that postoperatively (131,561).
The diagnosis of intermediate uveitis is based on the clinical findings. Absence of chorioretinal involvement, minimal anterior segment inflammation, and presence of vitreous exudates and pars plana exudates, which are pathognomonic for pars planitis, papillitis, and associated retinal phlebitis, suggest the diagnosis of intermediate uveitis. Intermediate uveitis may be associated with various noninfectious entities such as sarcoidosis, MS, and primary Sjogren syndrome or infectious diseases such as LD, Whipple disease, peripheral toxocariasis, toxoplasmosis, syphilis, TB, and human T-cell lymphotrophic virus type 1 (HTLV-1). Pars planitis refers to the subset of intermediate uveitis in which these findings are present in the absence of an associated infection or systemic disease. Laboratory and ancillary investigations are guided by a careful history, review of systems, and exam. Asking parents about a possible exposure of a child to ticks, skin changes, or arthritis may suggest the possibility of LD. Contact with cats, the presence constitutional symptoms, and adenopathy may suggest possible Bartonella infection. Fevers, night sweats, and fatigue may be associated with TB or sarcoidosis. In these cases, chest x-rays may be diagnostic. Gallium scan or chest computed tomography (CT) is done on patients with equivocal or negative results and may reveal subclinical sarcoidosis. Serologic testing may include ACE enzyme and lysozyme, which when elevated, may suggest a diagnosis of sarcoidosis. It is important to remember that in children, ACE levels are physiologically elevated, whereas previous treatment with systemic steroids may suppress ACE levels producing a false-normal value in a child with sarcoidosis. Furthermore, these indices are not specific for sarcoidosis with elevated ACE and lysozyme levels found in patients with other granulomatous disorders such as TB and leprosy. It is very important to exclude infectious causes of intermediate uveitis. Serologic tests for LD, catscratch disease, syphilis, and toxocariasis should be considered as suggested by the history and review of systems together with less common entities such as Whipple disease, HTLV-1, and Epstein-Barr virus (EBV).
Severe anterior uveitis treated previously with topical steroids may create confusion due to spillover of inflammatory cells into anterior vitreous, presenting a picture of an intermediate uveitis. In such cases, if the history is not typical for AAU, one may choose to observe the course of the disease. Inflammatory cellular aggregates in the vitreous are found rarely in iridocyclitis and never in pure iritis.
Sarcoidosis has been described in association with intermediate uveitis (101,102). Pulmonary sarcoidosis may precede the diagnosis of pars planitis, follow it, or be found concurrently. Upon comparison of the typical ocular findings seen in pars planitis, such as CME, optic nerve swelling, periphlebitis, and retrobulbar optic neuritis, the intermediate patients with sarcoid did not show significant differences from intermediate uveitis patients without sarcoid (132,133).
Although intermediate uveitis is often considered one of the most benign forms of uveitis, accurate statistics about the overall prognosis of patients with this disease are not available. Our experience suggests that early and aggressive treatment of intraocular inflammation associated with intermediate uveitis is more effective in preserving a good visual function than waiting until visual acuity has decreased. We use a modified stepladder approach to treatment of intermediate uveitis as initially proposed by Kaplan (134). After treatable infectious and noninfectious entities that may simulate or present a clinical picture of intermediate uveitis have been ruled out, we use the following regimen. The first step is administration of topical corticosteroids in the presence of anterior segment inflammation, together with regional corticosteroid injections (triamcinolone, 40 mg every 3 to 5 weeks). Periocular injections may be safely performed in the office in children as young as 10 years old with good cooperation. We limit the number of periocular steroid injections to no more than six. Step two is oral NSAIDs, if inflammation recurs following the third injection, and topical NSAIDs in the presence of CME. Step three is a short course of systemic corticosteroids for persistent inflammation in spite of previous steps. Prednisone is prescribed at a dose of 1 mg/kg/ day orally, with the initiation of tapering after 2 weeks of treatment, and guided by the clinical response. We limit the oral corticosteroid therapy to no more than 3 months of tapering regimen. In cases of recalcitrant inflammation, step four consists of systemic IMT versus therapeutic pars plana vitrectomy (PPV) with endolaser and/or cryopexy peripheral retinal ablation. It is not clear, however, whether vitrectomy should precede systemic IMT or whether all patients should receive preoperative IMT. Immunomodulatory drugs used in our practice include cyclosporine, methotrexate, and mycophenolate mofetil as a first choice. Azathioprine is the second choice, and cyclophosphamide and chlorambucil are the drugs of last resort due to their potential for significant toxicity. Clinical experience suggests promising efficacy of TNF-blocking agents (infliximab and adalimumab) in those with refractory inflammation. As intermediate uveitis, especially pars planitis, may be associated with an increased risk for the development of MS and that TNF-blocking agents may potentiate demyelinating disease and have been associated with the development of lymphoma in children, one must exercise caution with vigilant monitoring of pediatric patients with intermediate uveitis on long-term anti-TNF treatment.
A comprehensive review of the literature (135) and our clinical experience based on a prospective randomized pilot study of patients with intermediate uveitis (136) and that of a retrospective analysis of the role of vitrectomy on the management of refractory pediatric uveitis (137) suggest that PPV is clinically relevant in the management of noninfectious, posterior intraocular inflammation, particularly that associated with intermediate and pediatric uveitis, with attenuation of disease activity and the number of inflammatory recurrences, a reduction in CME, a diminished requirement for anti-inflammatory medications, and an improvement in vision in carefully selected patients. specifically, our cohort consisted of 28 eyes of 20 pediatric patients with active uveitis with or without medical therapy, followed for a mean of 13.5 months with diagnoses ranging from pars planitis, idiopathic pan uveitis, and JIA-associated iridocyclitis. Among these, six eyes had an associated retinal vasculitis. At the most recent follow-up, 96% of patients achieved inflammatory control with or without concomitant medical therapy including five of six patients with persistent retinal vasculitis. While there was only a modest reduction in the need for systemic IMT postoperatively, it should be noted that no eyes had adequate inflammatory control prior to surgery. Vitrectomy was thought to contribute to inflammatory control in patients with recalcitrant uveitis, and especially those with retinal vasculitis, which is known to require more aggressive therapy, possibly allowing lower doses and shorter duration of immunomodulatory medications. Mean best corrected visual acuity (BCVA) improved at all time points. Concurrent intraocular procedures included cataract extraction with or without IOL implantation, lens explantation, endolaser and cryotherapy, and intraocular injections of triamcinolone acetonide and bevacizumab. Intraoperative retinal tears were noted in two eyes undergoing 20-G PPV with the development of one retinal detachment at 1 week postop. Cataract progressed in four eyes with preop clear lenses within 6 months of surgery (137). Careful consideration of the relative risks and benefit s and judicious case selection for children undergoing PPV for this indication is paramount; those with poorly controlled inflammation despite extended therapy with IMT or biologic therapies being the most likely experience a therapeutic benefit from surgery. Nevertheless, the definitive role of PPV in the management of noninfectious intermediate, posterior, and panuveitis in adults and children is uncertain and will require prospective hypothesis–driven randomized controlled collaborative trials.
Sarcoidosis
Sarcoidosis is a chronic multisystem noncaseating granulomatous disease of unknown cause that occurs rarely in children. There are two presentations of sarcoidosis in children: early onset, <4 years of age, and late onset, between 8 and 15 years of age (138,139). Early-onset patients are primarily Caucasian, and pulmonary involvement is less common (35%), making it more difficult to establish the diagnosis, especially since articular involvement may mimic JIA (131,140). In contrast, older children and adolescents usually present with multisystem disease similar to that seen in adults with sarcoidosis. There is generally no familial history of sarcoidosis, and bilateral hilar lymphadenopathy or pulmonary involvement may be present on chest radiographs in 90% of cases. Eye and skin lesions occur in 30% of late-onset juvenile sarcoidosis. Since other diseases, including mycobacterial or fungal infection and berylliosis, can also produce noncaseating granulomas, the histologic diagnosis of sarcoidosis is made by exclusion. Vasculitis is a relatively unique complication associated with juvenile sarcoidosis.
Blau syndrome (aka familial juvenile systemic granulomatosis) is an autosomal dominant granulomatous disease of childhood, with clinical features almost identical to early-onset sarcoidosis with 100% phenotypic correspondence to mutations in the NOD2 gene (CARD15) (142,143). The disease susceptibility locus is on the pericentromeric region of chromosome 16 (144). Renal and hepatic granulomas have recently been described in this disease (145,146). The granuloma of the skin and synovial biopsy from the patient with Blau syndrome are identical to those seen in sarcoidosis (146).
Prevalence of Sarcoidosis: Sarcoidosis is rare in children, but a patient as young as 3 months old has been reported (147). A study from Denmark has estimated an annual incidence of two to three cases of sarcoidosis per million children (148). The prevalence of sarcoidosis is higher in females than in males, but the majority of reported pediatric cases are similar in the two genders (149). In the pediatric series reported from the southeastern United States, sarcoidosis had a higher incidence among African Americans (150,151). In children aged 4 years and younger with sarcoidosis, 7% to 28% is African Americans, whereas among children aged 8 to 15 years, the percentage of African Americans increases to 72% to 81% (138,152,153). Within the United States, approximately 80% of childhood cases have been reported primarily in Virginia, North Carolina, South Carolina, Arkansas, and Louisiana, suggesting that the southeastern and south central states are an endemic area for childhood sarcoidosis (151,153–155). The disorder is common in Scandinavian countries. In contrast, the disease is rare in India, Southeast Asia, New Zealand, and China (155).
Clinical Signs of Sarcoidosis: Early-onset juvenile sarcoidosis is unique in that it is characterized by the triad of skin eruptions, arthritis, and uveitis. This may mimic the symptoms of JIA without bilateral hilar lymphadenopathy. Intermittent fever and synovial swelling may further contribute to this masquerade. Early differentiation of sarcoidosis from JIA is important in planning treatment strategies and in counseling patients and families with skin changes serving to help differentiate these diseases at their onset. The rash seen in JIA is transient and comprised of pink macules, whereas that observed in sarcoidosis presents with variable erythema, papules, plaques, and ichthyosiform lesions (156). The cutaneous manifestations may be the earliest sign, occurring before joint or eye involvement, with papules, plaques, nodules, erythema nodosum, and hypo- or hyperpigmented areas. Lupus pernio is frequent in adults but rare in children (157). A skin biopsy should be performed to confirm the diagnosis. Sarcoid arthritis in children is characterized by painless, boggy synovial and tendon sheath effusions with mild limitation of motion, whereas in JIA, there is pain, limitation of movement, and destructive changes found on radiographs. Parotid gland enlargement is a frequent finding in children with sarcoidosis, especially the early-onset type.
Late-onset juvenile sarcoidosis presents similarly to that seen in adults—a multisystem disease with lymphadenopathy, hepatosplenomegaly, parotid fullness, and pulmonary involvement as well as generalized constitutional signs and symptoms (fever, anorexia, and malaise) (158,159).
A wide spectrum of systemic vasculitis in pediatric sarcoidosis has been reported, including leukocytoclastic vasculitis, vasculitis of small- to medium-sized vessels, and large-vessel vasculitis. Early-onset juvenile sarcoidosis patients should be carefully followed for development of vasculitis.
Neurologic dysfunction secondary to sarcoidosis is rare in children. Granulomas are most common in the basal area of the meninges and brain, causing seventh nerve palsy and hydrocephalus (160). Growth deficiency has been reported in association with brain MRI abnormalities, and hypothalamic infiltration can manifest as diabetes insipidus (161,162).
Renal involvement may be related to the presence of granuloma in the renal parenchyma or to hypercalciuria with nephrocalcinosis (162–166). Sarcoid liver granuloma may be present in up to 90% of patients, and liver enzymes are often mildly elevated in such instances. A needle biopsy may identify the lesions (167).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


