FIGURE 31.1 The orange-red elevated mass demonstrates the characteristic appearance of choroidal hemangiomas.
Most circumscribed choroidal hemangiomas are located in the postequatorial fundus (5,8). The posterior margin of the majority of these circumscribed lesions is located within two disc diameters of the optic disc or the fovea (8). Most of these lesions are <19 mm in diameter and have a mean elevation of 3 mm (8). Choroidal hemangiomas do not grow significantly over time, although a few cases of such tumor growth have been documented (9,10).
Subretinal fluid is commonly associated with this tumor and is responsible for the most common presenting symptoms (7). Circumscribed choroidal hemangiomas have been associated with choroidal neovascularization (CNV) (11). These lesions have also been reported in rare instances to resolve spontaneously, leaving only a chorioretinal scar.
Visual acuity in patients with choroidal hemangioma can be completely normal or may be decreased from a number of mechanisms. Vision loss and visual field defects associated with choroidal hemangioma may result from exudative retinal detachment, induced hyperopia, macular edema, retinal degeneration, loss of photoreceptors, chorioretinal adhesions, and cystoid degeneration.
Diagnostic Studies
The diagnosis of choroidal hemangioma is usually made based on ophthalmoscopic findings, although several ancillary studies can be used to help confirm the diagnosis. Ultrasonography shows high internal reflectivity on A-scan and acoustic solidity on B-scan images. Fluorescein angiography (FA) demonstrates early filling within large vascular channels constituting the tumor. Late frames show diffuse hyperfluorescence due to leakage of dye from the tumor surface (12a). Indocyanine green angiography (ICG) shows accumulation of dye early with subsequent washout later in the study (12b). ICG pattern may be highly diagnostic in challenging cases.
Differential Diagnosis
Despite the characteristic clinical features of choroidal hemangioma, this tumor can be challenging to distinguish from other choroidal tumors including amelanotic melanoma, choroidal osteoma, choroidal metastasis, or retinoblastoma. In addition, these lesions can mimic posterior scleritis or other inflammatory conditions.
Management
Treatment of choroidal hemangiomas is generally limited to those that are vision threatening, and treatment planning depends upon tumor size and location (13). Hemangiomas that do not affect vision and are not associated with subretinal fluid are followed by observation. These lesions are characteristically nonprogressive in nature.
Laser photocoagulation can be an important treatment and is indicated when the tumor causes loss of vision secondary to serous retinal detachment (8,13). Historically, focal lesions were treated with laser to cause resorption of subretinal fluid and readhesion of the neurosensory retina and retinal pigment epithelium (RPE). Subretinal fluid has been reported to resolve in 62% to 100% of cases following laser therapy (7).
Similarly, radiation therapy was frequently used in the treatment of choroidal hemangioma, including external beam radiation, plaque radiotherapy, or proton beam irradiation. Numerous reports have indicated that these treatments offer good results in terms of successful resolution of subretinal fluid (14–18). Shields et al. (7) concluded that plaque radiotherapy should be considered early in the treatment of choroidal hemangioma lesions that are resistant to laser treatment before chronic macular edema develops. They recommended that patients with the greatest risk for poor visual acuity be treated aggressively with external beam radiation or low-dose plaque radiotherapy. Metamorphopsia may improve significantly following brachytherapy.
Recent advances in treatment provide additional alternatives including photodynamic therapy (PDT). PDT is an especially efficacious, safe, noninvasive option for tumors located at the fovea (19–26) and can reduce subretinal fluid associated with choroidal hemangioma. Often only one cycle of laser is necessary to see a response. As a result, many experts now use PDT as first-line management in symptomatic patients.
Vision Rehabilitation
Because choroidal hemangioma is a benign tumor that does not undergo malignant transformation, patients with this lesion have an excellent prognosis for life. Visual prognosis, however, is significantly less positive. Despite the benign nature of these tumors, approximately 50% of affected patients have long-term visual acuity of 20/200 or worse (7). Final visual acuity is dependent upon numerous factors including initial visual acuity, failure of previous laser treatment, multiple quadrants of subretinal fluid, chronic submacular fluid, chronic cystoid macular edema, or chronic retinal pigment epithelial changes (7).
Children with choroidal hemangiomas face the additional problem of amblyopia in the affected eye. Amblyopia can result from refractive changes and induced hyperopia due to undetected tumors. Because early intervention can be effective in achieving resolution of subretinal fluid and restoration of visual function, prompt referral to an ophthalmologist with experience treating circumscribed choroidal hemangioma is especially important in children suspected of having this lesion.
Roles of Other Physicians and Health Care Providers
In select cases where radiation therapy is indicated, a consultation with a radiation oncologist is warranted. They can review the risks and benefits of radiation exposure associated with a plaque versus external techniques in a pediatric patient.
Ethical Considerations
Not applicable.
Prevalence
Medulloepithelioma is a rare embryonal tumor that arises from the medullary epithelium on the inner layer of the developing optic cup (27). Verhoeff (28) first described this tumor histopathologically as a teratoneuroma in 1904. Medulloepithelioma is classified into teratoid and nonteratoid forms. The nonteratoid form, otherwise known as a dictyoma, represents only medullary epithelial cells. In contrast, teratoid forms of this tumor contain heterotopic tissues including cartilage, skeletal muscle, and brain-like tissue (29). Either form can be benign or malignant (27,30,31). In terms of epidemiology, medulloepithelioma is a very rare tumor, the exact prevalence of which is unknown.
Intraocular medulloepithelioma often has a malignant histologic appearance. Extraocular extension with distant metastasis to the lungs, mediastinum, and lymph nodes has been documented (30,32). Broughton and Zimmerman (30) reported in their study of 56 cases that 66% were malignant and that of those, 30% were teratoid. Shields et al. (33) reported in their series that 90% were malignant and of those 56% were teratoid. Despite a malignant histologic appearance, the Broughton series, as well as others (31,33,34), suggest that distant metastases are rarely if ever seen in the absence of local extraocular extension.
Environmental Factors and Genetics
No reported environmental or genetic factors have been associated with medulloepithelioma.
Worldwide Impact
The worldwide epidemiology of this malignancy is unclear; this is a very rare malignancy. As with other intraocular tumors, delay in diagnosis, particularly in less developed countries, can result in metastasis and death.
Pathophysiology
Differentiation of the medullary epithelium that lines the optic cup normally produces the retinal photoreceptors, glial and neuronal tissues, nonpigmented ciliary epithelium, pigmented epithelium of the iris, muscles of the iris, and components of the vitreous.
Histopathologic studies demonstrate that medulloepithelioma contains elements that resemble the medullary epithelium, the optic cup and/or vessel, pigmented and nonpigmented ciliary epithelium, vitreous, or neuroglia (30). Medulloepitheliomas often contain heterotopic tissues such as cartilage, rhabdomyoblasts, skeletal muscle, and brain-like tissues (29). This feature results from the capacity of the primitive medullary epithelium to differentiate into a variety of neural and mesenchymal tissue types.
Clinical Symptoms and Signs
Medulloepithelioma characteristically presents in early childhood, at a mean age of 4 years, but these tumors can also be diagnosed in adults. This lesion can present as a ciliary body mass, or more rarely as a tumor of the optic nerve or retina (34–36) . Steinkuller and Font (37) concluded that medulloepithelioma should be considered a neoplasm of the eye and of the orbit.
Medulloepithelioma frequently presents with decreased vision and pain. Other common signs include leukocoria and a mass in the anterior chamber, iris, or ciliary body (30). One of the earliest signs may be a notch in the lens, “a lens coloboma,” in the quadrant of the tumor (38). This coloboma occurs because the embryonal tumor prevents normal zonular development in the associated lens quadrant.
Medulloepithelioma has the capacity to progress and to become locally invasive and destructive to surrounding intraocular tissues. Loss of vision occurs secondary to cataract, subluxed lens, neoplastic cyclitic membrane formation, or glaucoma (33). Other findings associated with this tumor include uveitis and retinal detachment (30). Iris neovascularization has also been reported as a manifestation of medulloepithelioma (33,39). Neovascular glaucoma discovered in a child with a normal fundus examination should raise suspicion for occult medulloepithelioma.
The presence of cysts within a tumor is highly suggestive of medulloepithelioma (33). Foci of cartilage, appearing as whitish opacities within the tumor, are also a characteristic finding in the teratoid form of medulloepithelioma (33).
Diagnostic Studies
The diagnosis of medulloepithelioma is primarily a clinical one, and the role of additional studies to confirm the diagnosis is not well established. A-scan ultrasonography has been reported to show irregular high internal reflectivity with areas of moderate reflectivity, and B-scan can demonstrate characteristic cysts within the tumor (40). Shields et al. (41) described dense echogenic areas in a medulloepithelioma that are similar in appearance to calcifications in retinoblastoma. These authors also described the tumor’s vascular pattern, demonstrating numerous leaking vessels, on FA (41).
Magnetic resonance imaging (MRI) of medulloepithelioma is hyperintense on T1-weighted images and hypointense on T2-weighted images, a result similar to that for malignant melanoma (42). Ultrasound biomicroscopy has recently emerged as a helpful diagnostic tool for evaluating tumors involving the anterior uveal tract; this approach may be particularly useful in demonstrating the multicystic appearance of medulloepithelioma (41,43).
Differential Diagnosis
Given the wide variety of presentations and possible locations for medulloepithelioma, the differential diagnosis is broad. Diagnoses that should be considered include congenital and inflammatory conditions, such as persistent hyperplastic primary vitreous (persistent fetal vasculature syndrome), pars planitis, or vascular malformations, and numerous tumors, including retinoblastoma, melanoma, melanocytoma, iridociliary cyst, rhabdomyosarcoma, neuroblastoma, or teratoma.
Management
Treatment of medulloepithelioma is surgical and often requires enucleation. Medulloepithelioma frequently recurs following local resection (31,41). Furthermore, these tumors are friable and difficult to remove surgically. Small, anteriorly located tumors may occasionally be followed by observation and/or treated with local resection. Given the tumor’s high risk for recurrence and the subsequent necessity for enucleation, as well as the difficulty of local resection, most authors recommend primary enucleation (31,33). Cases involving known extrascleral extension may require exenteration.
Neither local radiation nor chemotherapy has an established role in the primary treatment of medulloepithelioma. In cases involving risk for recurrence of the tumor, however, these methods may be used as adjunctive therapy.
Vision Rehabilitation
The visual outcome in patients diagnosed with medulloepithelioma is poor, and most patients with this disease lose the affected eye. Life expectancy, however, for patients with medulloepithelioma is surprisingly good, despite the malignant nature of the tumor. In the largest series of 56 patients with medulloepithelioma, four known tumor-associated deaths were reported. The most important prognostic feature was extraocular extension (30).
As with other intraocular tumors, early diagnosis is important in determining final clinical outcome. Unfortunately, diagnosis is often delayed in patients with medulloepithelioma; most of these patients experience a delay in surgical treatment for more than 1 year after the initial onset of symptoms (30).
As this tumor is unilateral and often treated with enucleation, the critical recommendation for patients following treatment is wearing of shatterproof lenses to protect the remaining eye.
Roles of Other Physicians and Health Care Providers
The diagnosis and management of these rare malignancies requires additional expertise from ocular pathologists and pediatric oncologists. These patients are best treated in a tertiary center with multidisciplinary expertise in pediatric solid tumors.
Ethical Considerations
Much of the developing world often lacks the multidisciplinary expertise to manage these rare tumors. Delay in diagnosis and limited access to specialty health care can lead to advanced orbital invasion and in the malignant form of the disease significant morbidity and mortality.
Prevalence
Choroidal osteoma is a benign choroidal tumor consisting of mature bone. This disease was first described clinically by Gass et al. (44) in 1978. Choroidal osteoma is likely a congenital lesion, and it often presents in infants, although the diagnosis may not be made until adulthood. Lesions of choroidal osteomas are more common in females and most often found adjacent to the optic nerve or located within the macula.
The exact incidence and prevalence of choroidal osteoma are unknown, although it has been suggested that these tumors frequently are asymptomatic, often are initially misdiagnosed, and are not rare (45).
Environmental Factors and Genetics
Choroidal osteoma has no known association with environmental factors. No genetic associations have been identified.
Worldwide Impact
Worldwide impact is unknown as many of these cases are thought to be asymptomatic. Cases with asymmetric visual acuity may not come to medical attention in many developing parts of the world.
Pathophysiology
Choroidal osteoma is composed of bony trabeculae with osteoblasts, osteocytes, and osteoclasts (44,46). Large, endothelial-lined, cavernous spaces are covered with small capillary blood vessels. The choriocapillaris can be altered and even destroyed in the involved areas. Overlying RPE can undergo degeneration.
The pathogenesis of choroidal osteoma is not known. One suggested mechanism is that a focal choroiditis may lead to calcification near the optic nerve (47). Another possibility is that the choroidal osteoma represents an osseous choristoma, wherein normal tissue is absent at the affected site (48). The lesion’s peripapillary location is typical of other developmental tumors described in this chapter, such as astrocytic hamartoma, combined retinal hamartoma, and retinal capillary hemangioma.
Intraocular calcification can be associated with trauma, long-standing retinal detachment, inflammation, abnormalities in calcium and phosphorus levels, and phthisis bulbi. Choroidal osteoma has been reported following intraocular inflammation (47,49). Serum calcium, phosphorus, and alkaline phosphatase levels are, however, found to be within the normal range in patients with choroidal osteoma.
Because females are more frequently diagnosed with choroidal osteoma than males, a hormonal role may be suggested in the pathogenesis; however, endocrine abnormalities have not been documented in these patients. A hereditary component in the pathogenesis is evident in some cases (50). Noble (50) speculated that a congenital, possibly inherited, defect in the choroid remains undetectable until osseous calcification is initiated through the influence of additional factors.
Clinical Symptoms and Signs
The location of choroidal osteoma dictates the patient’s presenting symptoms. Although this lesion may be diagnosed at any age, it is likely present at birth. Choroidal osteoma frequently remains asymptomatic and undetected, especially in the community setting (45). Once the fovea is involved by the tumor, with associated neovascularization and exudative retinal detachment, the patient can present with decreased vision, visual field defects, and metamorphopsia.
Choroidal osteoma lesions are characteristically elevated, yellow-orange masses. The tumors can appear mottled or spotted due to clumping of orange or brown pigment on their surface (51). Frequently, choroidal osteomas are solitary, although they occasionally occur as multiple lesions. Small distinctive tufts of vessels are often found on the tumor surface. These feeder vessels are best visualized on FA, and characteristically, they demonstrate no leakage (52).
Choroidal osteoma presents posteriorly, usually within the macula or adjacent to the optic nerve. Approximately 75% of cases are unilateral, with tumor dimensions of up to 22 mm in diameter and up to 2.5 mm in height (46).
Subretinal fluid can be present overlying choroidal osteoma. This presentation raises the suspicion of CNV with subsequent formation of subretinal fluid and/or hemorrhage (44,52). CNV can occur in approximately one-third of patients with choroidal osteoma (52).
Slow growth of choroidal osteoma occurs in approximately 50% of patients with these tumors. Growth is demonstrated either in an overall increase in size or in the formation of pseudopod projections extending out from the tumor’s central mass (53). Rapid growth in choroidal osteoma has been reported (54). Spontaneous involution of the tumor has also been documented (55).
Diagnostic Studies
Choroidal osteoma can be diagnosed clinically. Although the characteristic appearance of this lesion often serves to differentiate it from other tumors, diagnostic studies are frequently helpful.
Ultrasonography is particularly useful in differentiating choroidal osteoma from other lesions. A-scan ultrasonography demonstrates a high-intensity echo spike from the inner surface of the tumor, with decreased amplitudes of orbital soft tissue echoes posterior to the tumor. B-scan indicates a highly reflective choroidal mass with acoustic shadowing posterior to the tumor (46,52).
FA demonstrates early diffuse, irregular hyperfluorescence, with hyperfluorescence of the vascular tufts, and late staining. In the late frames, the spider-like vascular tufts may stand out in negative relief as hypofluorescent lines against a bright background (46,52). Choroidal neovascular membranes exhibit an early lacy pattern of hyperfluorescence with early leakage.
On computed tomography (CT) scan, choroidal osteoma demonstrates the same density as bone. On MRI, these tumors exhibit characteristics similar to ocular melanoma: hyperintense to vitreous on T1-weighted imaging and hypointense on T2-weighted imaging (56).
Differential Diagnosis
The differential diagnosis of choroidal osteoma includes other intraocular tumors, such as amelanotic choroidal nevi, choroidal melanoma, choroidal metastasis, and circumscribed choroidal hemangioma. Choroidal osteoma may appear similar to osseous metaplasia that can occur in other tumors, and this lesion should also be carefully distinguished from posterior scleritis or subretinal hemorrhage. Idiopathic sclerochoroidal calcifications should be included in the differential diagnosis for choroidal osteoma; however, the former are more often multiple, often bilateral, and are found typically along the superotemporal vascular arcades (51).
Linear nevus sebaceous syndrome presents with osseous choroidal choristoma, which should be carefully distinguished from choroidal hemangioma. This syndrome, however, also presents with characteristic systemic findings: midline facial linear nevus of Jadassohn, seizures, cognitive developmental deficits, and multiple eye findings including lipodermoids; colobomas of the lids, iris, and choroid; and choroidal calcification (57).
Management
Choroidal osteoma is typically not treated when the patient is asymptomatic. Periodic dilated fundus examinations and self-monitoring with the use of an Amsler grid can facilitate timely diagnosis in patients who develop choroidal neovascular membranes.
Regression of choroidal osteoma by argon laser photoablation has been described (58). Laser photocoagulation has been shown to have some success when used for treatment of a subfoveal neovascularization membrane when vision is threatened (45,59). Laser treatment can cause retinal anastomosis with both arteriolar and venular vessels within the tumor (45). Successful treatment of a RPE leak by focal laser has been reported (45). Use of PDT for CNV in choroidal osteoma has also been described with successful closure of the CNV in a patient who refused laser treatment (60).
Because the etiology of choroidal osteoma is unknown at this time, no preventive measures have been suggested.
Vision Rehabilitation
Visual loss to 20/200 or worse has been reported to occur by 20 years past diagnosis in approximately 60% of patients with choroidal osteoma (53). The major cause of vision loss is CNV. At 20 years from diagnosis, the risk of developing CNV is more than 50%. In one study, successful treatment of these membranes was reported in only 25% of patients (53). Hemorrhage associated with choroidal osteoma typically resolves but can cause a subretinal, vision-limiting disciform scar. The prognosis for life in these patients is the same as for the general population.
Severe visual loss can occur in bilateral cases. Low-vision rehabilitation with appropriate aids such as magnifiers and specialty lenses may be indicated.
Role of Other Physicians and Health Care Providers
In some instances a low-vision expert may be beneficial to address severe bilateral visual loss—often in advanced long-standing disease with CNV.
Ethical Considerations
Not applicable.
CONGENITAL HYPERTROPHY OF THE RETINAL PIGMENT EPITHELIUM
Prevalence
Congenital hypertrophy of the retinal pigment epithelium (CHRPE) was first described by Buettner (61) in 1975. Although this tumor is congenital, the diagnosis may be made at any age. CHRPE presents as solitary or multifocal lesions. Isolated CHRPE lesions are seen in the normal population. These hyperpigmented, isolated lesions are flat and circumscribed and represent congenital hypertrophy of the pigment epithelium with no primary involvement of the overlying retina. Multifocal lesions appear in two forms. One form of multifocal disease is unilateral and consists of lesions grouped to resemble animal footprints. These lesions are typically demonstrated in only one sector of the fundus and are sometimes referred to as “bear tracks,” or congenital grouped pigmentation of the retina (62). The second multifocal form of CHRPE is bilateral. The multifocal lesions in the bilateral form assume a more random distribution and are not grouped together in a single sector of the fundus (62,63) (Fig. 31.2). Bilateral multifocal CHRPE is associated with familial adenomatous polyposis (FAP) (64–73).
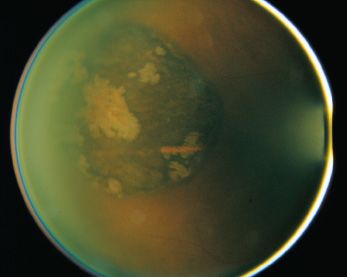
FIGURE 31.2 CHRPE epithelium, demonstrating characteristic lacunae.
FAP is a genetically inherited, autosomal dominant disease characterized by more than 100 polyps of the colon and rectum, predisposing these patients to colon cancer. Gardner syndrome involves the development of extracolonic manifestations of FAP and consists of intestinal polyps, characteristic patches of CHRPE, skeletal hamartoma, and soft tissue tumors. Both intracolonic and extracolonic presentations are now recognized as phenotypic variations of the same disease. CHRPE is the most common extraintestinal manifestation associated with FAP (74).
Because many CHRPE lesions may remain undiagnosed, the exact prevalence of this disease is unknown. FAP accounts for approximately 1% of all colon cancers, affecting 1 in 7,500 to 10,000 individuals (74).
Environmental Factors and Genetics
The gene responsible for FAP, designated the adenomatous polyposis coli (APC) gene, was cloned in 1991 and mapped to chromosome 5 (75,76). APC is a tumor suppressor gene, with a demonstrated involvement in the carcinogenesis of colon cancer (77). More than 250 APC mutations have already been identified (78).
This oncogene plays a role in tumor formation in the gastrointestinal tract, soft tissue, and bone. One study suggests that APC gene alterations may lead to defects in RPE melanogenesis and to focal RPE lesions (79).
Mutation analysis of APC is now an option for many families affected by FAP. A direct correlation has been demonstrated between the locus of the APC mutation and the retinal phenotypic disease expression. Careful delineation of the phenotype of the CHRPE lesions allows focused investigation of the APC mutation within certain coding regions. CHRPE lesions are only demonstrated when the mutation is located between codons 464 and 1,387 of the APC gene (80).
No environmental factors have been reported in association with CHRPE.
Worldwide Impact
None, as these lesions rarely affect vision and malignant transformation is rare.
Pathophysiology
Idiopathic solitary CHRPE histopathologically exhibits a monolayer of hypertrophied RPE cells with large pigment granules and photoreceptor degeneration overlying the RPE (61).
The histopathology described in Gardner syndrome demonstrates a more pervasive melanogenesis of the RPE (79). Several different configurations are described: a monolayer of hypertrophied cells, a mound of pigmented RPE cells between the RPE basement membrane and Bruch membrane, or a multilayered mound of hyperplastic RPE in a nodular or mushroom-shaped configuration (79,81). All of these conformations show cellular hyperplasia of the RPE. Although the fundus lesions in Gardner syndrome are referred to as CHRPE, this distinctive feature of cellular hyperplasia corresponds to a hamartomatous malformation of the RPE, not hypertrophy alone.
Electron microscopy has shown absence of autofluorescent lipofuscin granules in CHRPE lesions suggesting that RPE cells in CHRPE lack the catabolic functions of normal RPE cells (82).
Clinical Symptoms and Signs
Congenital hypertrophy of the RPE can present as a solitary lesion or as a collection of multiple lesions. Although some investigators classify these two disease forms differently, both forms are included in this discussion due to their similarities. Several of their clinical and histopathologic differences have previously been described in this section.
CHRPE lesions are congenital and have been observed in newborns. Solitary CHRPE lesions are demonstrated in the normal population as a benign incidental finding. A patient presenting with CHRPE typically has normal vision and normal anterior segments. Fundus examination reveals a flat, circumscribed, nonprogressive lesion. Size, shape, and degree of pigmentation of the lesions are quite variable (83). CHRPE lesions can be oval or round, with areas of pigmentation and depigmentation, and are often surrounded by a characteristic halo of depigmentation. CHRPE can present anywhere in the fundus; approximately 70% of these lesions are located in the temporal quadrant (84).
While CHRPE lesions are typically flat and nonprogressive, five patients were reported to have a nodular lesion arising from CHRPE (85). In each case, the nodular growth slowly progressed, fed by its own vasculature, causing exudative retinal detachment and chronic cystoid macular edema (85). This presentation may represent secondary reactive RPE proliferation or an acquired adenoma from CHRPE (85). One case of adenocarcinoma was reported to arise from CHRPE, suggesting that CHRPE lesions should be observed for neoplastic development, although this disease course is unusual (86).
The Association of Congenital Hypertrophy of the Retinal Pigment Epithelium with Gardner Syndrome and Familial Adenomatous Polyposis
FAP, Gardner syndrome, and CHRPE are all closely related. The ocular lesions in FAP have been reported to be bilateral in 86% of cases (83). Ocular lesions are observed in the presence or absence of other systemic manifestations of Gardner syndrome. The presence of multiple fundus lesions (more than four) or bilateral lesions has been reported to be a highly specific and sensitive phenotypic marker for Gardner syndrome (87). CHRPE lesions have been reported to be present in about two-thirds of families with FAP (83).
Families with FAP can differ in the presenting number and type of RPE lesions, although studies have demonstrated that affected individuals within a single family have similar pigmented lesions (88). In families with known FAP, the presence of retinal lesions revealed by fundus examination is highly predictive for the development of intestinal polyps; however, a negative examination cannot exclude risk for polyp formation (88).
Diagnostic Studies
CHRPE epithelium lesions are very distinctive and are generally diagnosed clinically. Studies, however, can support the diagnosis. FA demonstrates blockage of choroidal fluorescence in the hyperpigmented areas of the lesions in all phases of the study. Hypopigmented areas can exhibit hyperfluorescence, both early and late (window defect). CHRPE lesions do not leak on FA, as the overlying retinal vasculature and choriocapillaris are normal.
Visual field testing can indicate scotomas that correspond to the lesions, presumably representing progressive degeneration of the overlying photoreceptors (61). Because CHRPE lesions are flat, ultrasound studies may be unremarkable.
Differential Diagnosis
The differential diagnosis of a solitary CHRPE lesion should include choroidal nevus or choroidal melanoma. Multiple bilateral CHRPE lesions indicate greater suspicion of an underlying polyposis syndrome. Chorioretinal scars from toxoplasmosis, secondary hyperplasia of the RPE, sickle cell disease (sunburst lesions), sector retinitis pigmentosa, and pigmented retinopathies should also be considered in the differential diagnosis of CHRPE lesions.
Management
Treatment is not indicated for either solitary or multifocal CHRPE lesions. These lesions should be observed routinely, and if the patient’s medical history is suggestive, FAP or Gardner syndrome should be investigated. As rare malignant transformation has been described, an annual dilated examination is recommended.
Vision Rehabilitation and Prognosis
The prognoses for vision and for life are very good with solitary CHRPE lesions. In patients with FAP or Gardner syndrome, however, the prognosis for life is altered by the risk of malignant transformation of the colonic polyps. Ophthalmologic examinations revealing multiple bilateral CHRPE lesions may identify children and families at risk for developing polyposis and colon cancer. Rare malignant transformation of a solitary CHRPE to adenocarcinoma has been described.
Roles of Other Physicians and Health Care Providers
Patients with FAP or Gardner syndrome should be referred to a gastrointestinal specialist with expertise in managing colonic polyps. In some instances, surgical intervention and referral to an oncologist may be indicated.
Ethical Considerations
Not applicable.
COMBINED HAMARTOMA OF THE RETINA AND RETINAL PIGMENT EPITHELIUM
Prevalence
Combined hamartoma of the retina and of the RPE was first described by Gass (89) in 1973. Gass organized these lesions into categories based on location within the fundus: on the disc, next to the disc, in the macula, and in the periphery.
A hamartoma is a benign proliferation of cells that normally are found in the affected area. This ocular hamartomatous malformation involves the sensory retina, the RPE, the retinal vasculature, and the overlying vitreous. Combined hamartoma of the retina and RPE is a rare lesion, and its exact prevalence is unknown.
Environmental Factors and Genetics
No known environmental factors or genetic associations have been identified for combined hamartoma of the retina and RPE.
Worldwide Impact
Unknown, but likely very small as most cases are unilateral.
Pathophysiology
Combined hamartoma of the retina and RPE appears histopathologically with disorganized retina and retinal vascular tortuosity (90). Hyperplastic cells of the RPE migrate into the retina (91). Gliosis is present at the retinal surface leading to retinal distortion and folding. The vitreoretinal interface is altered with associated traction on the sensory retina.
These ocular hamartomatous lesions are characterized by benign growth of glial, vascular, or pigmented tissue from the RPE. Some lesions contain prominent vascular tissue while others are composed predominantly of glial tissue, which can lead to formation of preretinal or epiretinal membranes.
The pathogenesis of combined hamartoma of the RPE and retina is uncertain. Although the majority of patients with combined hamartoma do not present with other systemic diseases, these lesions have been reported in association with neurofibromatosis (NF), tuberous sclerosis (TS), incontinentia pigmenti, bilateral colobomas of the optic disc, optic nerve head drusen, juvenile retinoschisis, juvenile nasopharyngeal angiofibroma, Gorlin syndrome, and sickle cell anemia (89,92–96). These associations may indicate a developmental etiology, as suggested by Gass (92).
Clinical Symptoms and Signs
Combined hamartoma of the retina and RPE presents most frequently as painless loss of vision. Other presenting symptoms include metamorphopsia, floaters, strabismus, leukocoria, and occasionally ocular pain (92). Combined hamartoma may also be discovered as an incidental finding on routine ophthalmologic examination. Changes in vision associated with these lesions occur due to a variety of factors: involvement of the optic nerve or fovea, alterations at the vitreoretinal surface with epiretinal membrane formation and subsequent macular traction, or subretinal and intraretinal exudation.
On ophthalmologic examination, combined hamartoma of the retina and RPE appears pigmented and elevated, often with distortion of the retina and tortuous overlying vessels (Fig. 31.3). Because the pigmentation in these lesions is variable, the diagnosis is challenging in some cases. The location of the lesion, either juxtapapillary or peripheral, may also have an effect on its appearance (89). A juxtapapillary lesion characteristically appears as a solitary elevated mass adjacent to or immediately overlying the optic disc. In these lesions, contraction of overlying glial tissues often results in striae and distortion of the retina. Peripheral lesions can resemble an elevated ridge with accompanying traction of the vessels toward the lesion (97).
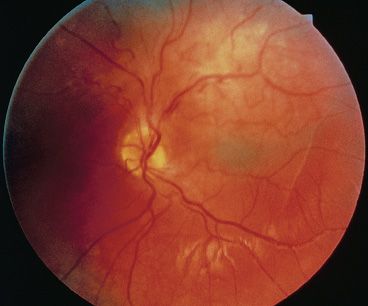
FIGURE 31.3 Combined hamartomas of the retina and RPE are pigmented elevated masses that are frequently associated with overlying glial tissues that produce retina distortion and striae.
Combined retina and RPE hamartoma is typically unilateral, although bilateral cases have been described (94,98). When bilateral, combined hamartoma is frequently associated with type 1 or type 2 NF (62,99–104). Ocular combined hamartoma is a congenital lesion. The diagnosis can be made at any age, including infancy and most frequently in childhood (89,92,105). Combined hamartoma of the retina and RPE is characteristically nonprogressive, although cases involving growth have been reported (106,107). Complications associated with combined retina and RPE hamartoma include retinoschisis, retinal holes, vitreous hemorrhage, CNV, retinal hemorrhages, and exudative retinal detachment (92,108,109).
Diagnostic Studies
FA is a useful tool in evaluating ocular combined hamartoma. Early FA phases demonstrate large tortuous vessels within the tumor, abnormal retinal capillaries, and late leakage of tumor vessels.
Ultrasound studies are not frequently useful in evaluating these lesions. Optical coherence tomography demonstrated an elevated hyperreflective mass, hyporeflective shadowing of the underlying tissues, and cystoid macular edema (110).
Differential Diagnosis
Combined hamartoma can be challenging to distinguish from an epiretinal membrane, particularly when the hamartoma is minimally pigmented and minimally elevated. Both hamartoma and membrane involve vitreoretinal interface changes associated with vascular tortuosity. Pigmentation and elevation are clues that the presentation is a hamartoma. Elevated and pigmented lesions can resemble choroidal melanoma, although the latter lesion does not have vitreoretinal interface changes or vascular tortuosity. Retinoblastoma or Toxocara canis may be considered initially in the differential diagnosis of lightly pigmented combined hamartomatous lesions in young children (111).
Peripapillary combined hamartoma has a presentation similar to morning glory disc anomaly, although the latter condition does not exhibit the characteristic elevation of combined hamartoma. With their tractional distortion of the retina, peripheral hamartomatous lesions can mimic retinopathy of prematurity.
Management
No well-established treatment plan has been developed for combined hamartoma of the retina and RPE. Therapy for this lesion has included treatment for amblyopia, which has demonstrated utility in selected patients (92). Vitreous surgery with epiretinal membrane removal is another treatment with reported success (112–114). Other patients with combined hamartoma have been treated with pars plana vitrectomy and membrane peeling without any subsequent improvement in their visual acuity (92,115).
Vision Rehabilitation
Patients with combined hamartoma of the retina and RPE demonstrate a wide spectrum of visual acuity. In one study, approximately one-quarter of the patients lost at least two lines of visual acuity; approximately one-third of patients exhibited vision of 20/200 or worse (92). A patient affected with the characteristically unilateral form of this disease typically experiences normal vision in the unaffected eye. No report has been made of malignant transformation of combined hamartoma of the retina and RPE lesions. The prognosis for life in these patients is normal.
In cases with extreme unilateral visual loss, amblyopia management may be considered.
Roles of Other Physicians and Health Care Providers
Not applicable.
Ethical Considerations
Not applicable.
The phakomatoses are a group of syndromes characterized by multiple associated lesions in multiple organ systems. Characteristically, patients with phakomatoses demonstrate hamartomatous malformations, which are abnormal proliferations of tissues that are normally found in the affected organ system. All these syndromes demonstrate ocular manifestations upon fundus examination. Phakomatoses discussed individually in this chapter include von Hippel-Lindau (VHL) syndrome, TS, NF, Wyburn-Mason syndrome (WMS), and SWS.
Von Hippel-Lindau Syndrome
Prevalence
VHL syndrome is an autosomal dominant condition characterized by angiomas of the retina, cerebellum, brain stem, and spine, accompanied by adenomas, angiomas, and cysts of the kidney, liver, and pancreas. Renal cell carcinoma occurs in approximately 25% of VHL patients, and pheochromocytoma occurs in approximately 10% (62). Incidence of VHL is estimated at 1 in 36,000 births (116).
Pathophysiology
Histopathologically, the retinal lesions of VHL are hemangioblastomas identical to the lesions also found in the central nervous system (CNS) (117). These vascular masses are composed of retinal capillaries exhibiting normal endothelium, basement membrane, and pericytes. Capillaries within the lesion may demonstrate abnormal fenestrations (118,119). Plump vacuolated interstitial cells with foamy cytoplasm, which are likely of glial origin, separate the capillary channels (117,120,121). Growth of these retinal lesions can extend inward toward the vitreous (endophytic type) or outward toward the choroid (exophytic type) (122,123).
Environmental Factors and Genetics
The pathogenesis of VHL disease is associated with a mutation in the VHL tumor suppressor gene (124). This syndrome follows an autosomal dominant inheritance pattern with variable penetrance. The VHL gene was mapped to chromosome 3p25 in 1988 and was cloned in 1993 (125,126). Recent studies have suggested that tumor formation in VHL disease follows the “two-hit” model initially hypothesized by Knudson (116,126–128) for retinoblastoma. According to this model, germline transmission of one mutation (first hit) is followed by a genetic alteration of the second allele in specific somatic tissues (second hit) (129). Loss of both normal functioning alleles of the VHL gene results in subsequent loss of functional VHL protein. Normal VHL protein appears to down-regulate production of vascular endothelial growth factor (VEGF) (130,131). Both the absence of functioning VHL gene product and the expected up-regulation of VEGF have been demonstrated in retinal capillary hemangioma (127). Mutations in the VHL gene are highly variable and include large deletions, small deletions or insertions, and nonsense or missense point mutations (124). Genetic diagnosis by direct mutation analysis may be possible in up to 75% of families with VHL (132–134) . DNA testing is a valuable tool in patients with retinal findings suggestive of VHL, particularly in families having no known history of the disease (135).
No specific environmental factors have been associated with VHL.
Clinical Features
Retinal hemangioblastoma is observed in approximately two-thirds of patients with VHL disease (136,137). Of reported VHL patients with retinal tumors, 25% will demonstrate associated cerebellar hemangioblastoma. Many other organs are affected by cysts, including the pancreas, kidneys, liver, adrenal glands, and epididymis. VHL is also characterized by renal cell carcinoma (22%), and pheochromocytoma is a less common but serious association (137). Although patients with VHL demonstrate brain and CNS tumor involvement, they exhibit normal cognitive capacity.
A patient with VHL disease may present with decreased visual acuity or may be asymptomatic with retinal tumors discovered as an incidental finding upon routine ophthalmologic examination. Alternatively, a patient may become symptomatic when retinal lesions cause a visual field defect associated with subretinal fluid, or retinal detachment; these lesions may also cause metamorphopsia from a macular pucker. Retinal lesions in VHL may become visible upon ophthalmologic examination during childhood. Because patients are frequently asymptomatic, however, they may be diagnosed at any time throughout adulthood.
In VHL disease, retinal capillary hemangioma appears as globular red-orange masses in the fundus. A characteristic feature of these retinal tumors is a pair of dilated, tortuous feeding and draining vessels traveling between the lesion and the optic nerve (Fig. 31.4). Angiomas may form anywhere in the fundus: rarely at the posterior pole (1%), more commonly at the optic disc (8%), and most frequently in the temporal peripheral retina (136).
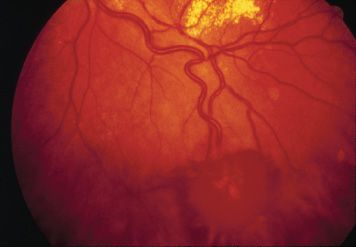
FIGURE 31.4 Retinal capillary hemangioma in VHL syndrome has a striking appearance. This example demonstrates a pair of tortuous feeder vessels.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


