FIGURE 30.1. The role of platelet-derived growth factor (PDGF)-B on blood vessel growth and mural cell coverage in a corneal neovascularization model. A: Endothelial cells were labeled by staining with lectin (green) and mural cells were stained with an antibody against smooth muscle actin (red). Starting at 10 days following corneal injury, mice received daily intraperitoneal injections of an anti–PDGFR-β antibody or phosphate-buffered saline (PBS) and were sacrificed at 20 days postinjury. Treatment with the anti–PDGF-β antibody led to reduced mural cell coverage compared to controls (arrows). Scale bar: 20 μm. B: Following induction of corneal injury, mice received daily intraperitoneal injections of one of the following: PBS, a polyethylene-glycolated anti-VEGF aptamer, an anti–PDGFR-β antibody, or both the anti-VEGF aptamer and the anti–PDGFR-β antibody. Neovasculature (green) was stained by fluorescein isothiocyanate-concanavalin A. Neovascularization was significantly reduced by the anti-VEGF aptamer compared with either PBS or the anti–PDGFR-β antibody (p < 0.01); inhibition of both VEGF and PDGF-B signaling led to a further significant reduction (p < 0.05) compared to inhibition of VEGF signaling alone. Scale bar: 100 μm. (Adapted from Jo, et al., Am J Pathol. 2006;168:2036–2053, with permission.)
MEDIATORS OF INFLAMMATION
The development of ocular neovascularization bears many of the hallmarks of an inflammatory process whose full nature is still being elucidated; this link appears to be relevant not only for AMD but also for other neovascular syndromes, such as retinopathy of prematurity (ROP) and DR. Several different types of data support the importance of inflammatory cells and factors classically associated with inflammatory processes. These include (a) both genetic and experimental data supporting a role of the complement cascade12,13; (b) the presence of macrophages in surgically excised choroidal neovascular membranes,14,15 together with the dependence of experimentally induced ocular neovascularization on the migration of macrophages (Fig. 30.2)16–19; (c) the clinical findings that an antibody against tumor necrosis factor-α (TNF-α), an inflammatory cytokine, can alleviate symptoms of AMD20; and (d) evidence that the influx of inflammatory cells is central to the attack on the retinal vasculature and the resulting ischemic neovascularization that are characteristic of DR.21
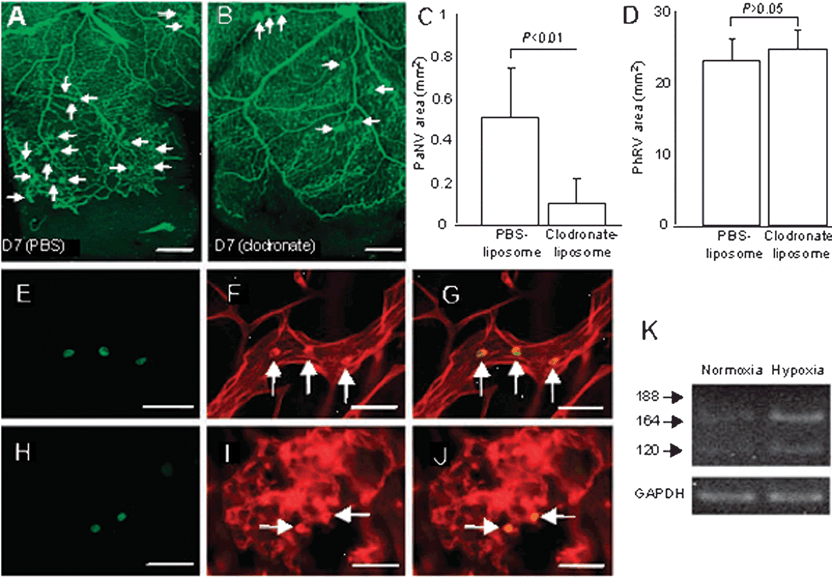
FIGURE 30.2. Monocytes contribute to pathological retinal neovascularization. In an ROP model, postnatal day zero (P0) rats were maintained for 10 days in 80% oxygen, interrupted daily by 30 minutes in room air, followed by a progressive return to 80% oxygen. This treatment led to an avascular retina. On P10, corresponding to study day 0 (D0), retinal revascularization was induced by maintaining the rats in room air for an additional 7 days (D7). A–C: At D7, pathological neovascularization (PaNV; arrows in A and B) was significantly inhibited by treatment with clodronate liposomes compared to control liposomes (n = 8 for both treatments; means ± standard deviation). D: Physiological neovascular area (PhRV) was not significantly affected by treatment with clodronate liposomes (p > 0.05). E–J: Influx of monocytes was observed just before and during pathological neovascularization (H–J). Monocytes were labeled with a fluorescein-conjugated antibody to CD13 (E and H), while rhodamine-conjugated concanavalin A was used to label the retinal vasculature and adherent leukocytes (F and I). As shown by superposition of these figures (G and J), the concanavalin A and CD13 staining colocalized, indicating that the adherent leukocytes were monocytes. K: In cultured peripheral blood monocytes obtained from retinopathologic rats at D7, exposure to hypoxia (1% oxygen) led to a marked increase in the expression of VEGF mRNA compared to exposure to normoxia (21% oxygen). Scale bars: (A and B) 0.5 mm and (E–J) 50 μm. (Reproduced from Ishida, et al. J Exp Med. 2003;198:483–489, with permission.)
Given the importance of anti-VEGF therapies for current treatment protocols, it is of particular interest that VEGF itself is known to act as a potent inflammatory cytokine in models of both ischemic neovascularization17 and DR-associated retinal pathology.21–23 The data supporting the importance of other inflammatory mediators in promoting ocular neovascularization offer the hope that they too may prove to be attractive therapeutic targets. Several of these mediators of inflammation are discussed in this section.
Complement Factors C3a, C5a, and CD59
Evidence supporting a role for the complement system in ocular neovascularization has come from both clinical and preclinical data. Genetic studies have identified specific haplotypes of complement factor H, a regulatory component of the complement cascade, as a significant risk factor for AMD.12,13 In addition, complement factors C3a and C5a have attracted particular interest as promoters of pathological ocular neovascularization. These factors accumulate in drusen of AMD patients, and this accumulation also is observed experimentally in mice after induction of CNV by laser wounding.24 Inhibition of experimental CNV is inhibited by the genetic ablation of the receptors for C3a and C5a24 or of C3 itself,25 as well as by complement depletion with cobra venom.25 As intravitreal injection of these factors was found to upregulate VEGF expression,24 the degree to which their induction of CNV is mediated through VEGF-independent pathways remains to be determined. A phase 1 trial (NCT00709527; Ophthotech) is underway to assess the utility of ARC1905, an aptamer against C5, given either alone or in combination with ranibizumab as a treatment for neovascular AMD. Another phase 1 trial is examining the effect of POT-4, an analog of the potent C3 inhibitor compstatin,26 as an intravitreal treatment for AMD (NCT00473928; Potentia, Pharmaceuticals, Louisville, KY).
Other studies of the molecular mechanisms of the complement system have found that CD59, a regulatory component, could directly inhibit neovascularization. When expressed from adenovirus-expressing transgene in a mouse model system, CD59 inhibited the deposition of membrane attack complexes on the retinal pigment epithelium (RPE).27 Direct administration of a CD59 fusion protein, either intravitreally or intraperitoneally, significantly inhibited the induction of CNV by laser wounding, while in the converse experiment, genetic ablation of CD59 enhanced the development of CNV.28
Tumor Necrosis Factor-α
TNF-α is a potent proinflammatory cytokine known to be involved in a variety of nonimmune regulatory functions as well.29,30 These include a role in promoting angiogenesis that may be mediated, at least in part, through upregulation of molecules such as VEGF31and its receptor, VEGF receptor-2 (VEGFR2),32 as well as PDGF-B.33 Preclinical studies in rodent models have demonstrated the efficacy of agents targeting TNF-α in reducing laser-induced CNV.34,35 In addition, two small case series reported that intravenous administration of infliximab, an anti–TNF-α monoclonal antibody, provided visual benefits for patients with neovascular AMD20 and diabetic macular edema.36 A small-scale study employing intravitreal injections of infliximab has also been encouraging,37 with the intravitreal approach being evaluated in a phase 1 trial involving patients with AMD and diabetic macular edema (NCT00695682; Retina Research Foundation, Houston, Texas).
Integrins
The integrins comprise a large family of membrane-bound cellular receptors, each consisting of an α and a β subunit, that mediate signaling through cell-cell contact or by interacting with components of the extracellular matrix.38 The α5β1 integrin is of particular interest as a therapeutic target, with small molecule inhibitors of the α5β1 integrin having demonstrated efficacy in preclinical studies of ocular neovascularization.39–41 These include JSM6427, which suppressed retinal neovascularization caused by ischemia39 as well as laser-induced CNV when administered systemically,40 and JSM5562, which inhibited corneal neovascularization.41 Intravitreal administration of JSM6427 also produced a dose-dependent reduction in clinically significant lesions in a laser-induced CNV model in monkeys without any related ocular adverse effects or evidence of systemic accumulation.42 JSM6427 is in a phase 1 trial (NCT00536016; Jerini AG, New York, NY) for the treatment of neovascular AMD. Volociximab, a monoclonal antibody that targets the α5β1 integrin, also was found to inhibit experimentally induced CNV in monkeys43 and is being evaluated in a phase 1 trial for the treatment of neovascular AMD (NCT00782093; Ophthotech).
Mammalian Target of Rapamycin
Mammalian target of rapamycin (mTOR) is a component of a heterotrimeric protein kinase that signals through two protein complexes, mTORC1 and mTORC2, to regulate a variety of processes, including angiogenesis44 and inflammation.45 Rapamycin inhibits the formation of mTORC144 and has been shown to reduce VEGF expression in RPE cells46 and to inhibit ocular angiogenesis in various preclinical models, including CNV as well as retinal and corneal neovascularization.47–49 Rapamycin also has been found to inhibit VEGF-induced vascular permeability.50 Similar findings with respect to ocular neovascularization and VEGF-induced vascular permeability have been reported for Palomid 529, another mTOR inhibitor that inhibits both mTORC1 and mTORC2 pathways.51–52
Clinical trials are investigating the utility of rapamycin for the treatment of neovascular AMD. In a phase 1 study of intravitreal or subconjunctival rapamycin, rapamycin was well tolerated and associated with improvements in both visual acuity and retinal thickness53; this study is being continued as a phase 2 trial (NCT00712491, MacuSight, Union City, CA). In addition, another phase 2 trial is examining the use of subconjunctival rapamycin in combination with intravitreal ranibizumab (NCT00766337; MacuSight), while oral rapamycin is being compared in a phase 2 trial (NCT00304954; National Eye Institute, Bethesda, Maryland) to two anti-inflammatory agents (infliximab and daclizumab) given intravenously; both of these trials are for the treatment of AMD.
Placental Growth Factor
Placental growth factor (PlGF) is a protein structurally related to VEGF that is also a ligand for VEGF receptor-1 (VEGFR1). PlGF is involved in a wide range of physiological processes54,55 such as recruitment of endothelial progenitor cells56 and modulation of immune system responses.57 PlGF has been implicated in pathological angiogenesis related to inflammation and ischemia,58 including laser-induced CNV.59 The role of PlGF in ocular angiogenesis may involve recruitment of monocytes60 that appear to be required for the maximal induction of ischemia-induced ocular neovascularization.22 VEGF-TRAP, a fusion protein containing the VEGF binding domains of VEGFR1 and VEGFR2 and that correspondingly targets both VEGF and PlGF, is currently in phase 3 trials (NCT00509795, Regeneron, Tarrytown, New York, NY; NCT00637377, Bayer, Leverkusen, Germany) for the treatment of neovascular AMD.
Pigment Epithelium-derived Factor
Pigment epithelium-derived factor (PEDF) is a secreted glycoprotein that acts as a natural inhibitor of angiogenesis61,62 and ocular inflammation.63 In many respects, it acts in direct opposition to VEGF, downregulating ischemia-induced VEGF expression in the retina64 and inhibiting VEGF-induced proliferation65 and endothelial permeability.66 In contrast to VEGF’s actions as an endothelial survival factor,67 PEDF induces endothelial cell apoptosis.68–70 In preclinical models of ischemia-induced retinal neovascularization, PEDF proved inhibitory whether administered intravitreally,65 systemically,68 or by expression from an intraocularly injected adenovirus.71 Some findings have proved contradictory, however, in that CNV induced by laser wounding was inhibited at low PEDF doses but actually increased at higher doses.72 The full elucidation of these dose-response issues will be required to permit further exploitation of PEDF as an inhibitor of CNV. To date, PEDF has been examined in one phase 1 trial (NCT00109499; Genvec, Gaithersburg, MA) in which an intravitreally administered adenovirus vector was used to express PEDF in patients with AMD with encouraging results.73
Renin/Angiotensin System
Evidence implicates the renin/angiotensin system in the development of inflammatory ocular neovascularization. In the classic renin/angiotensin pathway, the sequential action of renin and angiotensin-converting enzyme (ACE) ultimately yields angiotensin II, which then activates its type 1 and type 2 receptors.74 Blocking ACE activity was found to inhibit laser-induced CNV.75 Intraperitoneal injection of telmisartan, a blocker of the angiotensin II type 1 receptor (AT1-R), also has been found to inhibit both corneal neovascularization76 and laser-induced CNV,77 as well as ischemia-induced pathological neovascularization78; physiological retinal vascularization was not affected.78 As angiotensin II has been shown to upregulate VEGF expression79 while telmisartan reduced both leukocyte influx and expression of VEGFR1,78 it was suggested that blocking the AT1-R ultimately affected VEGF-mediated monocyte influx.78
In addition, an entirely different pathway involving the action of prorenin with its receptor may regulate ocular neovascularization in that administration of a decoy peptide to block this receptor led to inhibition of pathological neovascularization in the ischemic retinopathy model80 and of laser-induced CNV.81 Furthermore, the decoy peptide further inhibited CNV even if the AT1-R was ablated, showing that the actions of the decoy peptide were not simply a result of preventing the generation of angiotensin II.81
Angiopoietins
Angiopoietin-1 (Ang1) and angiopoietin-2 (Ang2) are both ligands for the Tie-2 receptor tyrosine kinase, through whose actions they regulate the development and remodeling of the vascular system.82 Usually, although not always, they act in an antagonistic manner, with Ang2 acting as a proinflammatory agent, destabilizing vascular endothelial cells and facilitating their response to transient signaling.83 In contrast, Ang1 provides a continuous stabilizing signal to the quiescent vasculature and in general exerts an anti-inflammatory action.83
These differences are exemplified in the interactions of Ang1 and Ang2 with VEGF. In transgenic model systems in which the expression of Ang1 and VEGF can be induced simultaneously, these two molecules act cooperatively in promoting angiogenesis. However, the vessels so induced are much less permeable than those induced by VEGF alone,84 owing to the inhibition by Ang1 of the VEGF-induced internalization of vascular endothelial cadherin, a key component of tight junctions VEGF.85 Similarly, overexpression of Ang1 inhibited the development of retinal neovascularization as well as CNV induced by laser wounding,86 suggesting that Ang1 could provide a therapeutic option for AMD.
In contrast, the interactions of Ang2 with VEGF tend to be destabilizing, with overexpression of Ang 2 furthering the promotion of endothelial cell permeability87 and of ocular neovascularization,88,89 although in the context of low levels of VEGF, Ang2 actually induced the regression of the neovascularization.89 This latter action has led to the suggestion that combinatorial therapy, in which an anti-VEGF agent is administered together with Ang2, might hold promise in treating CNV.90
Vascular Adhesion Protein-1
Vascular adhesion protein-1 (VAP-1) is a cell-surface glycoprotein expressed on endothelial cells that is involved in leukocyte recruitment during inflammation.91 In a recent study of laser-induced CNV in rats, VAP-1 was found to be upregulated and expressed in choroidal vessels; the blockade of VAP-1 by the injection of a specific VAP-1 inhibitor (U-V002) resulted in reduced CNV together with a lower expression of inflammatory markers such as TNF-α, monocyte chemoattractant protein-1, and intercellular adhesion molecule-1.92
ENDOGENOUS INHIBITORS OF ANGIOGENESIS
Investigations into the mechanisms mediating both physiological and pathological angiogenesis have identified a number of endogenous inhibitors, many of which are proteolytic fragments of larger molecules. The present discussion focuses on those for which a role in regulating ocular neovascularization has already been established. In contrast to molecules such as VEGF, for which therapeutic agents are used to block their action, these proteins are being examined as potential therapeutic agents in their own right.
Endostatin
Endostatin, a proteolytic fragment of collagen XVIII, is a potent endogenous inhibitor of angiogenesis.93,94 Endostatin is localized in Bruch membrane and in the choriocapillaris; patients with AMD show markedly reduced levels of immunoreactivity in these tissues.95 In a murine model of laser-induced CNV, the genetic ablation of collagen XVIII/endostatin led to significantly larger lesions, while in the converse experiment, intraperitoneal administration of endostatin in wild-type mice almost totally eliminated the formation of lesions.96 Similar reductions in laser-induced CNV were observed when endostatin was overexpressed from a transgene,97,98 and in another murine model, ischemia-induced retinal neovascularization was significantly reduced by intravitreally injected endostatin.99 Taken together, these findings suggest that endostatin, especially as an intravitreal agent, could provide a viable therapeutic option in treating AMD.
Angiostatin
Angiostatin, a name collectively given to various proteolytic fragments of plasminogen, is another endogenous inhibitor of angiogenesis,63,100 acting as a proapoptotic factor on endothelial cells while also inhibiting their migration and tube formation.101 Data very similar to those for endostatin suggest that angiostatin could prove a useful therapeutic agent. In a murine oxygen-induced retinopathy model, both systemic and intravitreal administration of angiostatin inhibited the pathological neovascularization without affecting physiological vascularization.102 In addition, both preretinal neovascularization induced by retinal vein occlusion103 and CNV induced by laser wounding98–104 were significantly inhibited by the expression of angiostatin from transgenes carried on viral vectors.
Thrombospondin-1
Thrombospondin-1 (TSP-1) is one of five related members of a glycoprotein family that is involved in physiological processes that include angiogenesis, wound healing, and blood coagulation.105 In angiogenesis, TSP-1 has been found to inhibit endothelial cell proliferation, adhesion, and migration106 and to promote endothelial cell apoptosis.107 It is synthesized by numerous ocular cell types,108 including the RPE,109 but, like endostatin, its presence has been detected in much lower amounts in the RPE and choriocapillaris of patients with AMD than in normal controls.95 In rodent preclinical models, TSP-1 significantly reduced ischemia-induced neovascularization whether injected intravitreally110 or when expressed from a transgene.111
VEGFxxxb Isoforms
VEGFxxxb denotes a family of VEGF isoforms that, compared to those that have been the focus of most investigations,112 have an altered carboxyl terminus owing to alternative splicing. As a result, these isoforms can bind VEGFR2 but do not initiate downstream signaling and thus serve as endogenous competitive inhibitors of VEGF.113 This inhibition has been demonstrated to have important physiological consequences. For example, the downregulation of VEGFxxxb has been correlated with the occurrence of malignancies.113 VEGFxxxb isoforms have been measured in the ocular fluids of human subjects; in diabetic patients with proliferative DR, they comprised only 12% of the total VEGF in the vitreous compared with 64% in control subjects, suggesting that the altered ratio contributed to the development of DR.114 Moreover, administration of recombinant VEGFxxxb was found to inhibit blood vessel growth in mouse models of corneal115 and retinal116 neovascularization. Taken together, these findings suggest that isoforms of the VEGFxxxb family may play an important role in regulating VEGF signaling and that the efficacy of anti-VEGF agents thus may depend on the local VEGF isoform expression pattern. As a naturally occurring inhibitor, the VEGFxxxb family ultimately could prove to be useful as therapeutic agents.
Tryptophanyl-tRNA Synthetase Fragment
Tryptophanyl-tRNA synthetase fragment (T2-TrpRS) is a natural cleavage product of tryptophanyl-tRNA synthetase,117 which was shown to inhibit both physiological retinal angiogenesis and VEGF-induced angiogenesis in murine models117; in an ROP model, intravitreal T2-TrpRS dramatically inhibited preretinal pathological tuft formation and facilitated physiological vascularization of the obliterated retinal vasculature.118 The mechanisms underlying these differing actions are unclear; one possible action as an antiangiogenic agent may be through binding to vascular-endothelial cadherin, a component of intercellular junctions between endothelial cells.119 Recently, a combination of T2-TrpRS plus an anti-VEGF aptamer and an αvβ3 integrin antagonist proved to be especially potent in the inhibition of neonatal retinal angiogenesis and in obliterating tumor vasculature in murine models120; in addition, the combination of T2-TrpRS and an anti-VEGF aptamer proved highly effective as an inhibitor of pathological neovascularization in an ROP model.120
NOVEL TARGETS FOR INHIBITION
In addition to the molecular targets that have emerged from focused investigations into the mechanisms underlying pathological angiogenesis, interesting candidates originally identified as acting in other physiological processes have since been found to play roles in regulating angiogenesis as well.
Erythropoietin
Aside from its established role in promoting erythropoiesis, erythropoietin also is important for neuroprotection121 and recently has been found to protect RPE cells against oxidative damage122,123 and to promote angiogenesis through recruitment of endothelial precursor cells and upregulation of VEGF.124 Studies also support the relevance of erythropoietin in mediating ocular neovascularization. In addition to clinical findings demonstrating elevated ocular levels of erythropoietin in conditions such as proliferative DR,125 preclinical studies with murine models have demonstrated reductions in retinal neovascularization using intravitreal injections of either a soluble erythropoietin receptor125 or a small interference RNA directed against erythropoietin.126 These studies suggest that interference with erythropoietin signaling may be a viable strategy for treating AMD, though clearly the protective actions of erythropoietin for both neurons and the RPE mandate careful monitoring of potential adverse events.
Nicotinic Acetylcholine Receptors
Nicotinic acetylcholine receptors are known principally as the mediators of synaptic transmission, yet they also have been found to regulate ocular neovascularization. In cultured cells, nicotine was found to stimulate endothelial cell proliferation, migration, and tube formation127 and to upregulate the expression of VEGF128 and VEGFR2.129 Orally administered nicotine increased the severity of laser-induced CNV,130,131 an effect mediated partly by the increased recruitment of bone marrow precursor cells.132 Additionally, subcutaneous administration of mecamylamine, an inhibitor of nicotinic acetylcholine receptors, blocked CNV induction (Fig. 30.3).131 Topical administration of mecamylamine as a treatment for AMD is being examined in a phase 2 trial (NCT00607750; CoMentis, South San Francisco, California).
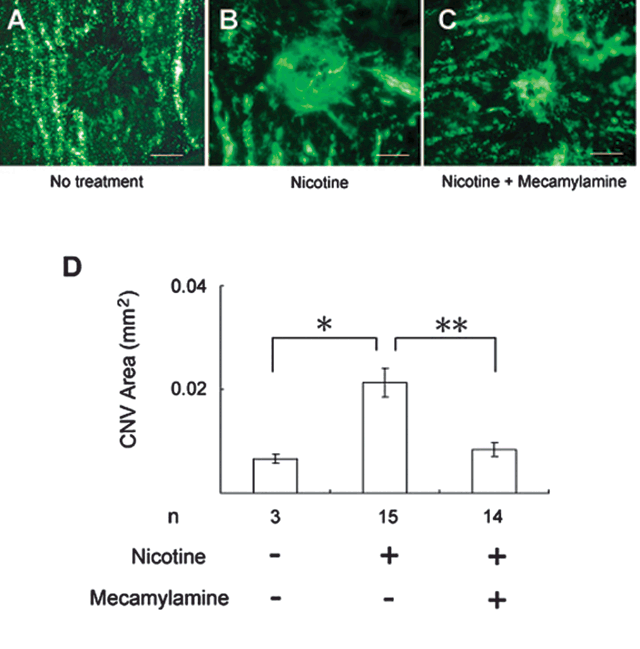
FIGURE 30.3. Mecamylamine blocks nicotine-induced stimulation of CNV. Six-week-old C57BL/6 mice were given 100 g/mL nicotine in their drinking water, and each underwent implantation of an osmotic pump that released mecamylamine or vehicle subcutaneously. After 2 days, these mice and a control group of three mice that did not receive any nicotine experienced a rupture of Bruch membrane at three locations in each eye. Fourteen days after laser, the mice were perfused with fluorescein-labeled dextran, and the area of CNV at Bruch membrane rupture sites was measured by image analysis. Compared with CNV lesions in untreated mice that did not receive nicotine (A), the lesions appeared larger in mice that received nicotine and underwent implantation of pumps that released only vehicle (B) but not in mice that received nicotine and mecamylamine (C). Measurement of the area of CNV by image analysis confirmed that nicotine caused a significant increase in lesion size that was completely blocked by mecamylamine (D). *p = 0.006470142; **p = 0.007367749; analysis of variance with Dunnett adjustment for multiple comparisons. (Reproduced from Kiuchi, et al. Invest Ophthalmol Vis Sci. 2008;49: 1705–1711, with permission).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


