FIGURE 30.1 A newborn infant with IP and stage 1 dermatologic manifestations. The vesicles and erythema are distributed linearly along the trunk and arms, sparing the face.
IP is a rare condition with approximately 1,000 cases reported in the literature (1). It does not appear to have an ethnic predilection, nor have any environmental factors been identified. Ophthalmic manifestations are reported in approximately 35% of patients with IP, though underdiagnosis of milder ophthalmologic manifestations may lead to an underestimation of the extent of ophthalmologic involvement (2). Approximately one in five IP patients will experience vision-threatening disease. When blindness occurs, it is often due to retinal detachment, which occurs in 3% of cases (2–4). Marked asymmetry of the retinal vascular disease is common.
Pedigrees of IP reflect classic mendelian inheritance in an X-linked dominant pattern. Because homozygosity is lethal in utero, only women inherit the disease. Though highly penetrant, the expressivity of the disease is quite variable, presumably due to female X chromosome inactivation (lyonization). De novo mutations appear to be common, as an affected pedigree is identified in only half of new cases (2). Affected women in such pedigrees often had male miscarriages. Extended pedigree analysis has demonstrated a 2:1 ratio of daughters to sons among affected mothers. Most reports of males with IP have no family history of the disease, suggesting somatic mosaicism (5). Rare reports of males with inherited IP can be explained by a Klinefelter karyotype (47,XXY), thus being exceptions that prove the rule (6). All males with the disease should have their karyotype evaluated for this reason. Appropriate genetic counseling should be offered to all affected families.
In 2000, the International Incontinentia Pigmenti Consortium identified mutations in the gene encoding nuclear factor kappa B(NF-kB) essential modulator (NEMO) as the cause of the disease (7). This small protein acts as a regulatory subunit for activation of NF-kB, a transcription factor that plays a critical role in the expression of genes crucial to cell survival and proliferation. NF-kB regulates genes involved in critical developmental processes, innate and adaptive immune responses, cell adhesion, and apoptosis. It also mediates inflammatory cascades (8). A genomic deletion accounts for 80% to 90% of the disease and suggests that the disease is caused by loss of function.
Indeed, targeted deletion of the gene encoding NEMO in mice recapitulates the human disease. NEMO-deficient male mice die in utero. The female mice develop patchy skin lesions characterized by granulocyte infiltration and hyperproliferation of keratocytes, similar to the histology of patients with IP (9).
Histopathologic studies of the eyes of NEMO-deficient mice have helped elucidate the pathogenesis of the retinal disease. Their retinal vasculature is characterized by increased arteriolar tortuosity. The arteriolar lumens are narrowed due to endothelial hypertrophy and basement membrane thickening (10). Complete retinal vasoocclusion is commonly observed in human histopathology of IP but was not seen in these mice (11). It seems plausible that the luminal narrowing represents an earlier stage of the disease evolution. Interestingly, the inflammatory changes and perivascular eosinophilic infiltration seen in dermatologic histology were not observed in the retinas of these mice.
The peripheral retinal perfusion of many patients with IP is abnormally reduced and appears to be responsible for much of the blinding complications of the disease. Fluorescein angiography of the disease reveals many similarities between the retinopathy of IP and retinopathy of prematurity (ROP). In both cases, vascular tortuosity is prominent. A rather abrupt transition occurs between mature vascular posterior retina and an avascular periphery. Posterior to the demarcation, arborizing arteriovenous anastomoses are present. In severe cases, later fibrovascular proliferation, exudation, and retinal detachment occur in a manner quite similar to ROP.
Several important findings distinguish the retinopathy of IP from ROP. Whereas the ischemia of ROP is entirely attributable to incomplete vascularization, retinal vasoocclusion of previously formed vessels appears to play a central role in the pathogenesis of IP. Fluorescein angiograms of the latter can reveal patchy areas of capillary dropout more reminiscent of diabetic retinopathy than the halted angiogenesis of ROP. Acute central retinal arterial occlusions have been reported in IP (12). While foveal hypoplasia appears common in children affected by ROP, the maculae of children with IP are characterized by an irregularly enlarged foveal avascular zone. The perifoveal capillary closure that characterizes this process appears to be a dynamic process. In some cases, abnormal vessels cross the ordinarily avascular zone of the fovea (12).
Children with IP can lose vision from disease of the retina, optic nerve, and brain (13). The severe vision loss of IP is often due to vitreoretinal complications of an avascular peripheral retina that can have a striking similarity to ROP. Vascular remodeling and collaterals demarcate an abrupt transition from vascular retina to avascular retina (Fig. 30.2). Often, epiretinal neovascularization develops at this border. In severe cases, this fibrovascular proliferation can lead to vitreous hemorrhage. It can also contract, leading to detachment of the neurosensory retina. The most severe cases develop funnel-shaped retinal detachment indistinguishable from the stage 4 and 5 ROP. Less severe cases can produce epiretinal neovascularization, temporal dragging, and macular heterotopias
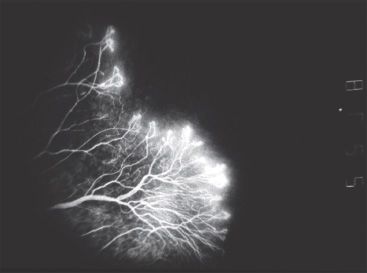
FIGURE 30.2 A fluorescein angiogram of a young girl with IP reveals peripheral nonperfusion, vascular loops, and preretinal neovascularization.
Macular pathology is another important cause of decreased vision among patients with IP. Studies with fluorescein angiography have revealed an absent or anomalous foveal avascular zone (Fig. 30.3). This has been termed “foveal hypoplasia.” Initially thought to reflect a developmental aberration, it more likely represents vascular remodeling after vasoocclusive events in the macula. An intriguing report of a transient classic “cherry-red spot” appearance, observed in a 12-day-old infant with IP, supports this hypothesis (14).
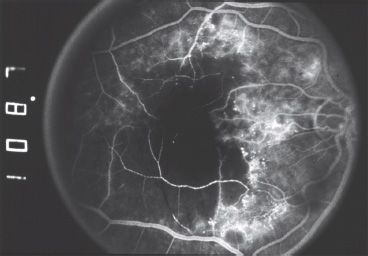
FIGURE 30.3 A fluorescein angiogram of a young girl with IP reveals multiple retinal arteriolar occlusions and capillary dropout, leading to a markedly enlarged foveal avascular zone.
Patients with IP can manifest abnormalities of the retinal pigment epithelium. These findings include increased macular pigmentation, subretinal pigmentary clumping, and mottling, sometimes associated with extensive epiretinal proliferation (proliferative vitreoretinopathy) (4,15). When these pigmentary changes are present, they are usually noted in the setting of significant retinal vascular disease, often complicated by vitreoretinal sequelae. These retinal pigment abnormalities likely reflect a secondary disease process (16).
Microphthalmos and congenital cataract have been described in 10% to 20% of large case series (17,18). Optic nerve atrophy also has been reported in these children. This atrophy likely reflects the extensive retinal damage that usually accompanies the finding, though some cases may be caused by intracranial pathology or possibly a primary vascular insult to the optic nerve.
Subepithelial corneal opacities can occur in children with IP, though there is only one report of such opacity appearing visually significant. In adults, the spectrum of anterior segment pathology that has been observed (cataract, iris atrophy, band keratopathy, and phthisis) is likely secondary to the underlying vitreoretinal pathology.
Strabismus is common among patients with IP. Though many of these cases can be attributed to sensory deprivation due to retinal or lens disease, intracranial pathology, anisometropia, and a positive kappa angle may be the cause in specific individuals
SYSTEMIC MANIFESTATIONS OF DISEASE
IP was originally recognized as a dermatologic disease. The widely adopted clinical criteria for diagnosing IP emphasize the dermatologic manifestations and do not account for CNS manifestations of the disease (Table 30.1) (19). These criteria were developed before identification of the genetic defect but still prove useful for establishing a diagnosis (1,17). However, genetically confirmed cases of IP without clinical dermatologic manifestations have been reported (19).
TABLE 30.1
Diagnostic criteria for IP
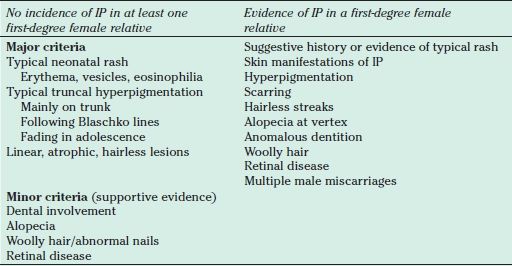
At least one major criterion is necessary to make a firm diagnosis of sporadic IP. The minor criteria, if present, will support the diagnosis, but because of their high incidence, complete absence should induce a degree of uncertainty.
IP, incontinentia pigmenti.
From Landy SJ, Donnai D. Incontinentia pigmenti (Bloch-Sulzberger syndrome). J Med Genet 1993;30(1):53–59.
The dermatologic disease can be described in distinct stages, though in any particular individual, the stages may overlap or be skipped (Fig. 30.1):
Stage 1: Diffuse blisters and erythema with relative sparing of the face
Stage 2: Hyperkeratotic papules develop on the distal limbs and scalp.
Stage 3: Linear hyperpigmented streaks and whorls along the lines of Blaschko
Stage 4: Pale, hairless patches often seen on the posterior calves
A transient mild dystrophy of the nails is common. Rarely, painful keratotic subungual tumors can develop (20). Mild alopecia is common, and the hair is often wiry and coarse (21).
Dental abnormalities are common (~80%). Though the enamel and overall tooth quality are normal, delayed eruption, hypodontia, and malformation lead to a typical dental appearance that is important to recognize and manage (20).
Breast abnormalities, including unilateral aplasia, hypoplasia, and supernumerary nipples, appear more common among patients with IP (19).
CENTRAL NERVOUS SYSTEM INVOLVEMENT
CNS abnormalities, together with retinal disease, account for most of the morbidity of IP. Large case series report that approximately 30% of patients with IP have clinically evident CNS disease (2). It appears that the CNS involvement is most common among those with prominent skin disease (22
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


