FIGURE 3.1 Vitreous from a 9-month-old child was dissected of the sclera, choroid, and retina. In spite of the specimen being on a surgical towel exposed to room air, the gel state is maintained. (Specimen is courtesy of the New England Eye Bank.)
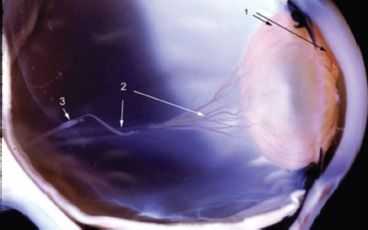
FIGURE 3.2 Human primary vitreous featuring the hyaloid artery (3) arising from the optic disc and branching to form the VHP (2), which anastomoses with the TVL and PM (1).
BIOCHEMISTRY OF THE VITREOUS BODY
At birth, vitreous is composed primarily of collagen resulting in a dense appearance on dark-field slit microscopy (Fig. 3.3), because collagen scatters light intensely. Hyaluronan (HA) synthesis begins after birth. Due to considerable hydrophilicity, HA generates a “swelling” pressure within the burgeoning vitreous that contributes to growth of the eye and also spreads apart the collagen fibrils to minimize light scattering inducing transparency (Figs. 3.1 and 3.4). This is evident when comparing dark-field microscopy of a human embryo (Fig. 3.3) with similar imaging in a 4-year-old child (Fig. 3.4) (see also Chapter 4).

FIGURE 3.3 Dark-field slit microscopy of vitreous from a 33-week-gestational-age human. The anterior segment is below where the posterior aspect of the lens can be seen. Coursing through the central vitreous is the remnant of the hyaloid artery, destined to become Cloquet canal. There is considerable light scattering by the gel vitreous.

FIGURE 3.4 Dark-field slit microscopy of vitreous from a 4-year-old child. The posterior aspect of the lens is seen below. Other than the peripheral vitreous cortex that contains densely packed collagen fibrils, there is little light scattering within the vitreous body.
There is heterogeneous distribution of collagen throughout the vitreous. Chemical (3,4) and light-scattering studies (5) have shown that the highest density of collagen fibrils is present in the vitreous base, followed by the posterior vitreous cortex anterior to the retina, and then the anterior vitreous cortex behind the posterior chamber and lens. The lowest density is found in the central vitreous and adjacent to the anterior cortical gel. HA molecules have a different distribution from collagen, being most abundant in the posterior cortical gel with a gradient of decreasing concentration centrally and anteriorly (6,7).
Both collagen and HA are synthesized during childhood. Total collagen content in the vitreous gel remains at about 0.05 mg until the third decade (8). As collagen does not appreciably increase during this time but the size of the vitreous does increase with growth, the density of collagen fibrils effectively decreases. This could potentially weaken the collagen network and destabilize the gel. However, since there is active synthesis of HA during this time, the dramatic increase in HA concentration may stabilize the thinning collagen network (9).
Bishop (10) proposed that the leucine-rich repeat protein, opticin, is the predominant structural protein in short-range spacing of collagen fibrils. Scott (11) and Mayne et al. (12) claimed that HA plays a pivotal role in stabilizing the vitreous gel via long-range spacing. However, studies (13) using HA lyase to digest vitreous HA found that the gel structure was not destroyed, suggesting that HA is not essential for the maintenance of vitreous gel stability, leading to the proposal that collagen alone is responsible for the gel state of vitreous (10).
PRIMARY VITREOUS
Vitreous vascularization begins with the hyaloid artery entering the intraocular space through the optic cup. By 10 weeks of gestation (WG), the hyaloid system is well established, branching to form the vasa hyaloidea propria (VHP) with anastomoses to a dense capillary network, the tunica vasculosa lentis (TVL), which is posterior to the lens, and the pupillary membrane (PM) adherent to the anterior surface of the lens and iris diaphragm (14). Maximal development of the hyaloid vasculature is reached by 12 to 13 WG (15) although evidence of regression is apparent as early as 11 WG (16). The hyaloid system shows clear signs of regression by 13 to 15 WG beginning in the VHP followed by the TVL and then the PM (16–18).
Although the precise biochemical process involved in hyaloid vasculature formation and regression is poorly defined, studies have shown that vascular endothelial growth factor (VEGF) may trigger the growth of the TVL and PM (19–22). Thus, decreased VEGF levels may induce vascular regression. Transforming growth factor (TGF) has also been implicated in ocular vasculature remodeling (23). Since all four forms of TGF-β were found in vitreous hyalocytes, these cells may be involved in the induction of hyaloid vessel regression. Other mechanisms may be involved. There is evidence that family members of the Wnt signaling pathway, particularly Wnt7b expressed in macrophages, are involved in normal hyaloid regression (24) in the mouse, although this mechanism has not been confirmed in humans.
SECONDARY VITREOUS
Secondary (avascular) vitreous formation arises from a remodeling of the earlier primary (vascular) vitreous (25). By the 40- to 60-mm stage in humans, the VHP has reached maximum maturity and regression begins (22). The posterior VHP is the first component to degenerate. The regression continues to move centrally affecting the TVL and finally the hyaloid artery itself. Although flow through the hyaloid artery is decreasing during this time, ceasing entirely around the 240-mm stage, the artery may continue to elongate as the eye lengthens (26). As the primary vitreous regresses, its remnants provide a framework for collagen fibrils of the secondary vitreous that form via interactive remodeling. As this occurs, the primary and secondary vitreous coexist in the same space during this transitional developmental period (25,26).
VITREOUS STRUCTURE
In the human embryo, vitreous structure (27–33) has a dense homogenous appearance (Fig. 3.3) primarily as a result of the aforementioned collagen synthesis during secondary vitreous formation. HA synthesis after birth separates collagen fibrils, inducing transparency (Fig. 3.4). In the absence of diabetes and myopia, vitreous remains largely transparent until around the fifth decade when fine, parallel fibers appear coursing in an anteroposterior direction (Fig. 3.5B and C) (2,30–32). The fibers arise from the vitreous base (Fig. 3.5H) where they insert anterior and posterior to the ora serrata. As the peripheral fibers course posteriorly, they are circumferential with the vitreous cortex, while central fibers “undulate” in a configuration parallel with Cloquet canal (33). The fibers are continuous and do not branch. Posteriorly, these fibers insert into the vitreous cortex (Fig. 3.5E and F).
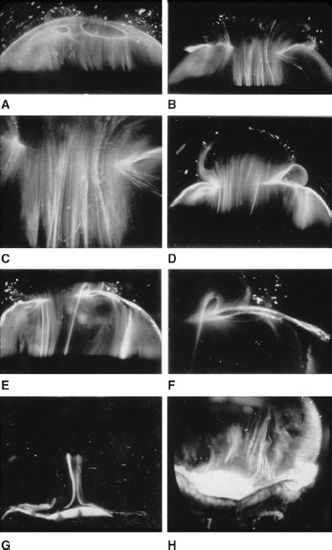
FIGURE 3.5 Dark-field slit microscopy of human vitreous in eyes dissected of the sclera, choroid, and retina. The anterior segment is below and the posterior pole is above in all images. (Courtesy of the New York Eye Bank for Sight Restoration.)
Ultrastructural studies (34) have demonstrated that collagen fibrils are the only microscopic structures that could correspond to these fibers. These studies also detected the presence of bundles of packed, parallel collagen fibrils. Eventually, the aggregates of collagen fibrils attain sufficiently large proportions so as to be visualized in vitro (Fig. 3.5) and clinically. The areas adjacent to these large fibers have a low density of collagen fibrils separated by HA molecules and therefore do not scatter light as intensely as the bundles of collagen fibrils.
Vitreous Base
Vitreous base is a three-dimensional zone extending 1.5 to 2 mm anterior to the ora serrata, 1 to 3 mm posterior to the ora serrata (35), and several millimeters into the vitreous body itself (36). The posterior extent of the posterior border of the vitreous base varies with age (37,38). In a variety of vitreoretinal developmental disorders, the temporal vitreous never forms; thus, there is a different peripheral terminus bounded by vitreous gel posteriorly and liquid vitreous anteriorly (see below). Vitreous fibers enter the vitreous base by splaying out to insert anterior and posterior to the ora serrata (Fig. 3.5H). The anterior-most fibers form the “anterior loop” of the vitreous base, a structure that is important in the pathophysiology of anterior proliferative vitreo-retinopathy (2,39–41). Studies by Gloor and Daicker (42) showed that cords of vitreous collagen insert into gaps between the neuroglia of the peripheral retina. They likened this structure to Velcro (a self-adhesive nylon material) and proposed that this would explain the strong vitreoretinal adhesion at this site. In the anterior vitreous base, fibrils interdigitate with a reticular complex of fibrillar basement membrane material between the crevices of the nonpigmented ciliary epithelium (43). The vitreous base also contains intact cells that are fibroblast-like anterior to the ora serrata and macrophage-like posteriorly. Damaged cells in different stages of involution and fragments of basal laminae, presumed to be remnants of the embryonic hyaloid vascular system, are also present in the vitreous base (43).
Vitreous Cortex
The vitreous cortex is the peripheral shell of the vitreous body that courses forward and inward from the anterior vitreous base to form the anterior vitreous cortex and posteriorly from the posterior border of the vitreous base to form the posterior vitreous cortex. The anterior vitreous cortex, often referred to as the “anterior hyaloid face,” begins about 1.5 mm anterior to the ora serrata. The posterior vitreous cortex is 100 to 110 µm thick (44) and consists of densely packed collagen fibrils (Fig. 3.6, bottom). There is no vitreous cortex over the optic disc (Fig. 3.4A), and the cortex is thin over the macula due to rarefaction of the collagen fibrils (44). The prepapillary hole in the vitreous cortex can sometimes be visualized clinically when the posterior vitreous is detached from the retina. If peripapillary glial tissue is torn away during posterior vitreous detachment (PVD) and remains attached to the vitreous cortex around the prepapillary hole, it is referred to as Vogt or Weiss ring. Vitreous can extrude through the prepapillary hole in the vitreous cortex (Fig. 3.4A) but does so to a much lesser extent than through the premacular vitreous cortex where it can produce traction and certain forms of maculopathy (45–47).
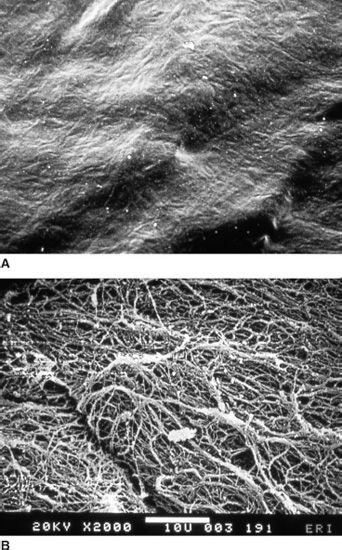
FIGURE 3.6 Scanning electron microscopy of the anterior surface of the ILL of the retina (top photo) and the posterior aspect of posterior vitreous cortex in a human.
Hyalocytes
Hyalocytes are mononuclear cells (Fig. 3.7) that are embedded in the vitreous cortex and widely spread apart in a monolayer 20 to 50 µm from the retina. Quantitative studies of cell density in the bovine (48) and rabbit (49) vitreous found the highest density of hyalocytes in the vitreous base, followed by the posterior pole, and the lowest density at the equator. Hyalocytes are oval or spindle-shaped, are 10 to 15 µm in diameter, and contain a lobulated nucleus, a well-developed Golgi complex, smooth and rough endoplasmic reticula, and many large periodic acid-Schiff–positive lysosomal granules and phagosomes (44,50). Balazs (51) pointed out that hyalocytes are located in the region of highest HA concentration and suggested that these cells are responsible for vitreous HA synthesis. There is evidence to suggest that hyalocytes maintain ongoing synthesis and metabolism of glycoproteins within the vitreous (52,53). Hyalocytes have also been shown to synthesize vitreous collagen (54) and enzymes (55).
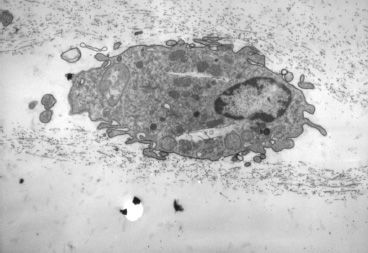
FIGURE 3.7 Transmission electron micrograph of human hyalocyte embedded in the posterior vitreous cortex characterized by the dense matrix of collagen fibrils. (Original magnification x11,670).
The phagocytic capacity of hyalocytes has been described in vivo (56) and demonstrated in vitro (57,58), consistent with the presence of pinocytic vesicles and phagosomes (50) and surface receptors that bind immunoglobulin G and complement (58). Hyalocytes become phagocytic cells in response to inducting stimuli and inflammation. HA may have a regulatory effect on hyalocyte phagocytic activity (59,60). As an extension of this phagocytic activity, hyalocytes can be antigenpresenting cells that modulate intraocular inflammation while also keeping the vitreous clear via phagocytic activity. Finally, collagen membranes with hyalocytes are known to contract in the presence of certain stimulants, such as TGF-β2. This phenomenon may explain the contraction of preretinal cortical vitreous responsible for a myriad of pathologies with preretinal membranes, especially macular pucker (61).
Vitreoretinal Interface
In the child, it is virtually impossible to mechanically detach the posterior cortical vitreous from the retina, resulting in surgical complications such as retinal breaks. The interface between vitreous and retina consists of a complex formed by the posterior vitreous cortex and the internal limiting lamina (ILL), which includes the basal lamina of Müller cells (62,63) (Fig. 3.6) (see also Chapter 4). The ILL is composed of type IV collagen closely associated with glycoproteins (64–66). Immediately adjacent to the Müller cells is the lamina rara, which is 0.03 to 0.06 µm thick and demonstrates no species variations nor changes with topography or age. The lamina densa is thinnest at the fovea (0.01 to 0.02 µm). It is thicker elsewhere in the posterior pole (0.5 to 3.2 µm) than at the equator or vitreous base. At the rim of the optic nerve head, the retinal ILL ceases, although the basement membrane continues as the inner limiting membrane of Elschnig (67). This membrane is 50 nm thick and is believed to be the basal lamina of the astroglia in the optic nerve head. At the central-most portion of the optic disc, the membrane thins to 20 nm, follows the irregularities of the underlying cells of the optic nerve head, and is composed only of glycosaminoglycans and no collagen (68). This structure is known as the central meniscus of Kuhnt. Balazs (7) has stated that the Müller cell’s basal lamina prevents the passage of cells as well as molecules larger than 15 to 20 nm. Consequently, the thinness and chemical composition of the central meniscus of Kuhnt and the membrane of Elschnig may account for, among other effects, the frequency with which abnormal cell proliferation arises from or near the optic nerve head (39,41).
Zimmerman and Straatsma (69) claimed that there are fine, fibrillar attachments between the posterior vitreous cortex and the ILL and proposed that this was the source for the strong adhesion between vitreous and retina. The composition of these fibrillar structures is not known, and their presence has never been confirmed. It is more likely that an extracellular matrix “glue” exists between the vitreous and retina in a fascial as opposed to focal apposition (30,64,65), comprised of fibronectin, laminin, and other extracellular matrix components (70). Western blots of separated proteins derived from dissected vitreoretinal tissues identified two chondroitin sulfate–containing proteoglycans of approximately 240-kDa molecular weight (MW) that are believed to function as adhesive molecules at the vitreoretinal interface. These findings formed the rationale for experimental and clinical studies on pharmacologic vitreolysis (71) using ABC chondroitinase (see Chapter 2.2).
DEVELOPMENTAL ABNORMALITIES OF VITREOUS
Development of the Embryonic Hyaloid Vasculature
Many developmental vitreoretinal disorders result from abnormalities in the embryonic vascular system of the vitreous (VHP) and lens (TVL). These attain maximum prominence during the 9th week of gestation or 40-mm stage (72). Hyaloid vessel development around the seventh WG appears to be driven by VEGF165 (an isoform of VEGF-A). However, once the hyaloid vascular system reaches completion, the alternatively spliced antiangiogenic VEGF165b isoform begins to dominate (73).
Regression of Fetal Vasculature
Atrophy of the vessels begins posteriorly with dropout of the VHP, followed by the TVL. Recent studies have detected the onset of apoptosis in the endothelial cells of the TVL as early as day 17.5 in the mouse embryo (19). At the 240-mm stage (7th month) in the human, blood flow in the hyaloid artery ceases. Regression of the vessel itself begins with glycogen and lipid deposition in the endothelial cells and pericytes of the hyaloid vessels (74). Endothelial cell processes then fill the lumen, and macrophages form a plug that occludes the vessel. The cells in the vessel wall then undergo necrosis and are phagocytized by mononuclear phagocytes (75) identified as hyalocytes (76).
Recent studies suggest that the VHP and the TVL regress via apoptosis (77). Mitchell et al. (19) point out that the first event in hyaloid vessel regression is endothelial cell apoptosis and propose that lens development separates the fetal vasculature from VEGF-producing cells, decreasing the levels of this survival factor for vascular endothelium, inducing apoptosis. Following endothelial cell apoptosis, there is loss of capillary integrity, leakage of erythrocytes into the vitreous, and phagocytosis of apoptotic endothelium by hyalocytes. Meeson et al. (78) proposed that there are actually two forms of apoptosis that are important in regression of the fetal vitreous vasculature. The first (“initiating apoptosis”) results from macrophage induction of apoptosis in a single endothelial cell of an otherwise healthy capillary segment with normal blood flow. The isolated dying endothelial cells project into the capillary lumen and interfere with blood flow. This stimulates synchronous apoptosis of downstream endothelial cells (“secondary apoptosis”) and ultimately obliteration of the vasculature. Removal of the apoptotic vessels is achieved by hyalocytes.
Proteomic analysis of embryonic (aged 14 to 20 WG) human vitreous (vitreomics) revealed that during hyaloid vessel regression there is a significant decrease in profilin-1 actin-binding protein and significant increases in cadherin-2 cell adhesion protein, cystatin-C protease inhibitor, dystroglycan cell adhesion molecule, clusterin, as well as pigment epithelium–derived factor (PEDF), both known to have antiangiogenic infuences (79). The presence of dystroglycan and profilin-1 was confirmed in the hyaloid vessels of 10, 14, and 18 WG embryonic human eyes by immuno-labeling (80). Clusterin was not present in the HA or TVL of 10 and 14 WG eyes, but was found in the HA of 18 WG eyes. Cadherin was not found in the hyaloid vessels of the 10 WG eyes but was observed in the hyaloid vessels at 14 and 18 WG (80).
Comparison of the protein composition of the adult human and developing human vitreous from the second trimester (14 to 20 WG) revealed 314 and 1,213 proteins, respectively, including 78 proteins that were unique to the adult and 1,002 unique to the fetal vitreous (81). A large fraction of the fetal vitreous proteins are intracellular and may be involved in the remodeling of the hyaloid vasculature and retina during the second trimester. Proteins that were found in greater abundance in fetal vitreous compared to adult vitreous included low MW kininogen-1, cystatin-B, insulin-like growth factor–binding protein (IGFBP)-2 and IGFBP-4, thrombospondin-1 and thrombospondin-4, thioredoxin, mu-crystallin, alpha-2-antiplasmin, nidogen-2 and growth differentiation factor 8, as well as secreted proteins pigment epithelial derived factor (PEDF), AMBP (α-1-mikroglobulin), hemoglobin, and Ig chains. Low MW extracellular matrix proteins detected in fetal vitreous included COL5A1, 6A3, 11A1, 15A1, 1A1, 2A1, 4A2, and 18A1 (81).
Bioinformatic analysis of the protein profile of fetal (14 to 20 WG) human vitreous revealed that most proteins were members of either the free radical scavenging, molecular transport, or small molecule biochemistry, or connective tissue disorders networks. Those proteins involved in free radical scavenging, molecular transport, and small molecule biochemistry were all intracellular and decreased during the second trimester. In contrast, the proteins in the connective tissue disorders network were all extracellular and increased during the second trimester. This pattern is consistent with replacement of the cellular primary vitreous with the acellular collagenous secondary vitreous. This analysis further revealed that EIF2 signaling and protein ubiquitination were the top canonical pathways (82).
These studies, essential because they are in humans, often lack comparison or controls, so mechanisms remain unclear and more research in this area is needed.
Pathologies of the Primary Vitreous
Persistence of the hyaloid vascular system occurs in 3% of full-term infants but in 95% of premature infants (83), 90% of infants born earlier than 36 WG, and in over 95% of infants weighing <5 pounds at birth (84). There is a spectrum of disorders resulting from persistence of the fetal vasculature (85), which can at times be associated with prepapillary hemorrhage (86).
a. Mittendorf dot is a remnant of the anterior fetal vascular system located at the former site of anastomosis of the hyaloid artery and TVL. It Is usually inferonasal to the posterior pole of the lens and is not associated with any known dysfunction.
b. Bergmeister papilla is the occluded remnant of the posterior portion of the hyaloid artery with associated glial tissue. It appears as a gray, linear structure anterior to the optic disc and adjacent retina and does not cause any known functional disorders. Exaggerated forms can present as prepapillary veils.
c. Vitreous cysts are generally benign lesions that are found in eyes with abnormal regression of the anterior (87) or posterior (88) hyaloid vascular system, otherwise normal eyes (89,90), and eyes with coexisting ocular disease, such as retinitis pigmentosa (91) and uveitis (92). Some vitreous cysts contain remnants of the hyaloid vascular system (93), supporting the concept that the cysts result from abnormal regression of these embryonic vessels (94). However, one histologic analysis of aspirated material from a vitreous cyst purportedly revealed cells from the retinal pigment epithelium (95). Vitreous cysts are generally not symptomatic and thus do not require surgical intervention. However, there is a report (96) describing the use of Nd:YAG laser therapy to rupture a free-floating posterior vitreous cyst.
d. Persistent fetal vasculature (PFV) (persistent hyperplastic primary vitreous, PHPV) was first described by Reese (97) and subsequently renamed PFV (85). Though there is a broad spectrum of presentations, PHPV/PFV often features a plaque of retrolental fibrovascular connective tissue adherent and posterior to the posterior lens capsule. The membrane extends laterally to attach to the ciliary processes, which are elongated and displaced centrally. Although 90% of cases are unilateral, many of the fellow eyes had Mittendorf dot or another anomaly of anterior vitreous development (98). A persistent hyaloid artery, often still perfused with blood, arises from the posterior aspect of the retrolental plaque in the affected eye. In severe forms, there can be microphthalmos with anterior displacement of the lens–iris diaphragm, shallowing of the anterior chamber, and secondary glaucoma. PHPV/PFV is believed to arise from abnormal regression and hyperplasia of the primary vitreous (97). Experimental data suggest that the abnormality begins at the 17-mm stage of embryonic development (99). The hyperplastic features result from proliferation of retinal astrocytes and a separate component of glial hyperplasia arising from the optic nerve head (100). The fibrous component of the PHPV/PFV membrane is presumably synthesized by these cells (101). A recent case report found that collagen fibrils in this fibrous tissue had diameters of 40 to 50 nm with a cross-striation periodicity of 65 nm. The investigators concluded that the collagen fibrils differed from those of the primary vitreous and suggested that they arose either from a different population of cells or were the result of abnormal metabolism by the same cells that synthesize vitreous collagen (102).
The retina is usually not involved in anterior PHPV, suggesting that the anterior form is due to a primary defect in lens development and that vitreous changes are all secondary (103). In posterior PHPV/PFV opaque connective tissue arises from Bergmeister papilla and persistent hyaloid vessels (101,104). These can cause congenital falciform folds of the retina and, if severe, can cause tent-like retinal folds, leading on rare occasions to tractional and/or rhegmatogenous retinal detachment. Font and investigators (105) demonstrated the presence of adipose tissue, smooth muscle, and cartilage within the retrolental plaque and suggested that PHPV/PFV arises from metaplasia of mesenchymal elements in the primary vitreous.
e. Familial (Dominant) Exudative Vitreoretinopathy Familial exudative vitreoretinopathy (FEVR) was first described in 1969 by Criswick and Schepens (106) as a bilateral, slowly progressive abnormality that resembles retinopathy of prematurity (ROP), but without a history of prematurity or postnatal oxygen administration (see also Chapter 28 on FEVR). Gow and Oliver (107) identified this disorder as an autosomal dominant condition with complete penetrance. They characterized the course of this disease in stages ranging from PVD with snowflake opacities (stage I) to thickened vitreous membranes and elevated fibrovascular scars (stage II) and vitreous fibrosis with subretinal and intraretinal exudates, ultimately developing retinal detachment due to fibrovascular proliferation arising from neo-vascularization in the temporal periphery (stage III). Plager et al. (108) recently reported the same findings in four generations of three families but found X-linked inheritance. van Nouhuys (109–111) studied 101 affected members in 16 Dutch pedigrees and five patients with sporadic manifestations. He found that the incidence of retinal detachment was 21%, with all but one case occurring prior to the age of 30. These were all tractional or combined traction–rhegmatogenous detachments, and there were no cases of exudative retinal detachment. van Nouhuys (109) concluded that the etiology of FEVR lies in premature arrest of development in the retinal vasculature, since the earliest findings in these patients were nonperfusion of the peripheral temporal retina with stretched retinal blood vessels and shunting with vascular leakage. Thus, van Nouhuys considers FEVR as a retinopathy with secondary vitreous involvement. However, Brockhurst et al. (112) reported that vitreous membrane formation began just posterior to the ora serrata and that it preceded retinal vessel abnormalities, suggesting a vitreous origin to this disorder. Others suggested that there may be a combined etiology involving anomalies of the hyaloid vascular system, primary vitreous, and retino-vascular dysgenesis (113).
f. Retinopathy of Prematurity: First described in the 1940s (114), retinopathy of prematurity remains a leading cause of blindness in children in the United States (115). Vitreous liquefaction, often unidentified (116) in stages 1 and 2 ROP, likely occurs as a result of reactive oxygen species (117) but also overlying the peripheral retina where immature Müller cells may not support typical gel synthesis and may account for the vitreous trough apparent during surgery for stage 4 ROP. The disrupted composition may limit the inherent vitreous ability to inhibit cell invasion (118–120), thereby permitting neovascularization in stage 3 ROP to grow (121,122) between posterior gel and peripheral liquid vitreous anteriorly (Fig. 3.8) (122). Instability at the interface between gel and liquid vitreous exerts traction on the underlying ridge. As ROP advances from stage 3 to 4, the neovascular tissue grows through the vitreous along the walls of the future Cloquet canal or the tractus hyaloideus of Eisner toward Wiegert ligament on the posterior lens capsule (123). Primary vitreous cells, responding to intraocular angiogenic stimuli, likely proliferate and migrate to help create the dense central vitreous stalk and retrolental membrane seen in the cicatricial stages.

FIGURE 3.8 Photomicrograph of peripheral fundus in retinopathy of prematurity. The lens is in the upper right hand corner. Below, a fibrovascular membrane is present at the interface between the posterior gel vitreous and the peripheral liquid vitreous. The inset shows the histopathology that clearly distinguishes between gel (G) and liquid (L) vitreous. (Courtesy of Maurice Landers, MD.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


