Pathophysiology
Mark E. Kleinman • Jayakrishna Ambati
Introduction
The chorioretinal architecture is a heterogeneous assembly of specialized tissues derived from neuroectoderm and mesenchyme. The dual-layered blood-retina barrier, which incorporates the retinal pigment epithelium (RPE), serves as a sentry for the vital cellular circuitry that integrates visual data while the underlying choroid provides the hypermetabolic photoreceptor segments with one of the richest nutrient exchange networks in the human body. In multiple inflammatory, infectious, and immune-mediated diseases, these tissues undergo specific cellular transformations, for which nature intended to protect and repair the choroid and retina, but which paradoxically lead to blindness. This chapter addresses the clinical and molecular pathogenesis of choroidal neovascularization (CNV) with a particular emphasis on critical growth factors and newly discovered cell signaling pathways that will yield an armamentarium of targeted therapeutics in the 21st century.
The choroid is a highly vascularized pigmented tissue layer engineered to provide efficient nutrient and metabolic exchange for the RPE and outer two thirds of the retina. In the setting of choroidal inflammation or injury, cellular components of the RPE and choroid activate in order to contain the wound, destroy any invading pathogens, and initiate the repair process. As a result of the subsequent proinflammatory surge, newly formed blood vessels invade the wound bed to revascularize hypoxic areas and provide growth factors to the damaged cells. Typically, as in several other peripheral tissues in the human body such as skin, this neovascular response allows for tissue healing and scar remodeling; however, in the delicate architecture of the chorioretinal interface, these abnormal vessels grow in an unregulated fashion beneath the retina and are susceptible to leakage and rupture. CNV and the resultant extravascular leakage of fluid into the subretinal space are hardly suitable for the highly specialized function of photoreceptors and lead to significant loss of visual quality. Through centuries of clinical observations and scientific investigation, critical insights into the biology of CNV in injury and repair have modernized our current understanding of the cellular and molecular pathways that drive this pathological process.
Pathogenic Mechanisms of CNV in Age-Related Macular Degeneration
The most common forms of CNV are due to a combination of inflammation, oxidative stress, and photochemical damage secondary to age-related macular degeneration (AMD), which is the leading cause of blindness among the elderly on three continents and affects 30 to 50 million people worldwide.1–3 CNV is a public health epidemic as its prevalence (~2 million) and annual incidence (~200,000) are as great as those of breast or prostate cancer in the United States.4,5 Between 10% and 20% of people with AMD will progress to the neovascular form, which is responsible for about 90% of vision loss in patients with AMD. Recent evidence suggests that the gradual accumulation of inflammatory debris in drusen beneath the RPE and within the choroid ultimately leads to the production of proangiogenic cytokines and retinal invasion by abnormal and leaky vasculature.6 Some of these inflammatory components are comprised of photochemically altered proteins that are linked to intermediate or terminal products of the vitamin A cycle or lipid breakdown products released by dying photoreceptors and RPE.7 There is now sufficient scientific evidence that drusen do not simply precede neovascular AMD but in fact promote pathologic angiogenesis in the setting of AMD. CNV can wreak havoc on visual acuity via leakage into the subretinal, intraretinal, or vitreous spaces near the macula. Overtime, these CNV membranes remodel into fibrotic scars that may still harbor abnormal vasculature and be sites of recurrently active neovascularization (NV).
The original diagnostic classification of neovascular AMD presented by Gass described two patterns of CNV as determined by fluorescein angiography and histopathologic examination (Fig. 3.1).8 Type I or occult CNV was characterized by late leakage of fluorescein, suggesting that the neovascular lesion was contained beneath the RPE layer. In Type II or classic CNV, fluorescein angiography demonstrates early, lacey hyperfluorescence and subsequent pooling in the subretinal space (Fig. 3.2). In these lesions, the CNV has grown through Bruch membrane and the RPE layer into the subretinal space. More recently, a third type of CNV termed retinal angiomatous proliferation (RAP) was described in which a pathologic anastomosis develops between the choroidal and retinal circulation via CNV growth through the RPE/Bruch membrane complex.9 These lesions usually occur later in the remodeling stages of CNV but are frequently noted on detailed angiographic studies. Another pattern of CNV, polypoidal choroidal vasculopathy (PCV), has also been associated with AMD and is currently an area of active investigation.10
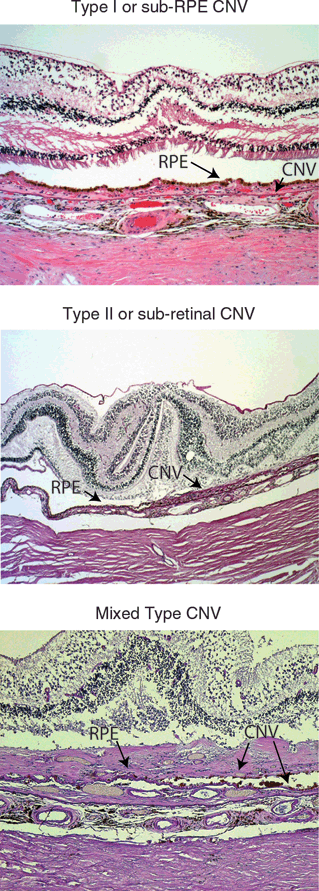
FIGURE 3.1. Histopathology of AMD-related CNV. Light micrographs of tissue sections from eyes with neovascular AMD demonstrated sub-RPE, subretinal, and mixed forms of CNV formation (hematoxylin and eosin). (Images courtesy of Dr. Hans Grossniklaus.)
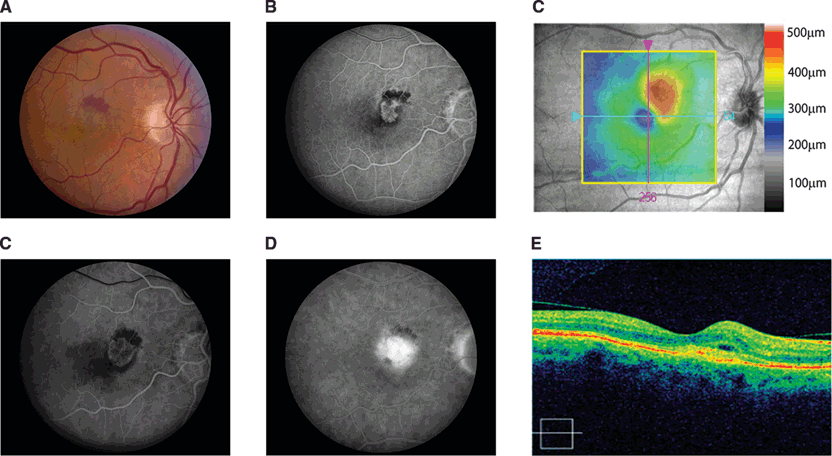
FIGURE 3.2. CNV formation in human eye with AMD. A: Color fundus photograph of patient with AMD and acute decrease in vision showed large CNV membrane with subretinal heme and fluid. B: Fluorescein angiogram of same eye at 18 seconds exhibited early lacey hyperfluorescence with large semicircular frond of predominantly classic CNV. C: Summed voxel projection image of retina on SD-OCT with significant retinal elevation in juxtafoveal area. D: An early fluorescein angiogram (21 seconds) from the same eye acquired 10 days later demonstrated the rapid growth of the CNV membrane. A late image revealed significant fluorescein leakage with pooling consistent with features of a predominantly classic CNV membrane. E: SD-OCT of the same eye confirmed the presence of subretinal fluid and disruption of the RPE. (Images courtesy of Drs John Kitchens and Scott Westhouse.)
Other Pathogenic Mechanisms of CNV
CNV may be caused by a wide-range of mechanical, cellular, physiologic, photochemical, and iatrogenic processes that disrupt the chorioretinal interface and lead to inflammation of the posterior segment. Each of these instigating factors, either individually or in concert with one another, is capable of setting off the intricate cascade of factors leading to CNV. While AMD is the most common etiology of CNV, below is a partial list of some of the other disorders that may also result in CNV formation:
Structural and Inflammatory Choroidal Diseases
There are several ophthalmologic disorders in which fractures develop within Bruch membrane such as angioid streaks and pathologic myopia, which allow for the in-growth of choroidal vasculature, subsequent NV, and leakage into the subretinal space. Presumed ocular histoplasmosis syndrome (POHS) is hypothesized to result from choroidal seeding of Histoplasma capsulatum, a fungal organism endemic to the Ohio and Mississippi River Valleys. Infected choroidal tissue undergoes cycles of inflammation and quiescence leading to localized areas of CNV, which eventually scar over with RPE and glial cells.
Choroidopathies are a group of rare disorders that affect the tissues around the RPE due to either immune system dysregulation or infectious disease. Multifocal choroiditis (MFC), multiple evanescent white-dot syndrome (MEWDS), bird-shot choroidopathy, serpiginous choroidopathy, and diffuse unilateral subacute neuroretinitis (DUSN) have all been associated with CNV, which is hypothesized to be secondary to chronic inflammation or immune dysregulation.
Laser-Induced Injury of RPE and Bruch Membrane
The use of laser photocoagulation for targeted thermal injury in the treatment of extrafoveal CNV and proliferative diabetic retinopathy (DR) has been widely used with success for several decades. In the treatment of CNV, focal laser injury is able to inhibit growth and decrease leakage from abnormal microvasculature. With panretinal photocoagulation, laser is used to ablate large swaths of the peripheral retina in DR that would otherwise be ischemic, producing proangiogenic factors. It is through the destruction of this pathologic tissue that NV is spared in the precious visual real estate in the macula. Laser-injured tissues will commonly remodel into small areas of RPE hyperpigmentation with fibrosis and only go on to develop CNV when high-energy laser photocoagulation ruptures Bruch membrane. It is critical to note that laser-induced injury, which fractures Bruch membrane, serves as a very well-described animal model of CNV, which is addressed in the following section.
Mechanical Trauma
Blunt trauma to the eye disorganizes the retinal architecture to varying degree. Choroidal rupture may induce neovascular invasion of the subretinal space via fractures in Bruch membrane. Over time, these lesions eventually involute but do leave behind a residual fibrotic scar that significantly decreases visual acuity in the area.
Animal Model of Laser-Injury–Induced CNV
In order to study the natural history of CNV in vivo, several experimental models11–13 have been developed to mimic the disease process that occurs in humans. However, it is the laser injury model of CNV that is the longest standing and most widely used experimental model of CNV and is accepted as a surrogate, accelerated model of neovascular AMD. Over 25 years ago, Stephen Ryan described the laser-induced injury model of CNV in rhesus monkeys.14,15 The same technology has since been used to develop similar models in a variety of research animals, including the mouse and rat. In this model, high-energy laser photocoagulation is used to fracture Bruch membrane, which results in the formation of CNV (Fig. 3.3). The laser-induced model captures many of the important features of the human condition including migration of choroidal endothelial cells into the subretinal space via defects in Bruch membrane, accumulation of subretinal fluid, congregation of leukocytes adjacent to neovascular tufts, gliosis, leakage of fluorescein from immature new vessels into the subretinal space, and increased expression of angiogenic growth factors and their receptors in cells and ocular tissues. This model has provided a powerful system in which we can dissect the molecular mechanisms of CNV formation and test potential targeted therapeutics.
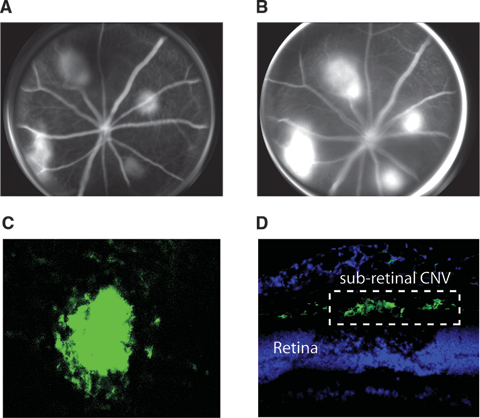
FIGURE 3.3. Laser-induced mouse model of CNV. A: An early time-point fundus fluorescein angiogram 7 days after laser-injury of the retina, RPE, and Bruch membrane showing hyperfluorescent areas. B: Later time-point fluorescein angiogram of the same mouse eye exhibiting significant fluorescein leakage similar to the human form of CNV. C: Choroidal flatmount of laser-induced CNV stained with fluorescent lectin to identify blood vessels showing a dense vascular lesion in choroid. Z-stacks of these entire lesions are acquired in order to measure the total CNV volume in preclinical therapeutic studies. D: Immunofluorescent microscopy of laser-injured mouse eyes stained with the endothelial cell marker, CD31 (green), reveals robust subretinal CNV. Nuclei are shown in blue.
VEGF-A is a Critical Mediator in the Pathogenesis in Neovascular AMD
The growth factor hypothesized as the “Factor X” in angiogenesis some 60 years ago was isolated and characterized as vascular endothelial growth factor (VEGF) in 1989.14–18 VEGF belongs to the highly conserved platelet-derived growth factor (PDGF) family along with several other related proteins that constitute the VEGF subfamily including placental growth factor (PlGF), VEGF-B, VEGF-C, and VEGF-D. For nomenclature purposes, the original VEGF protein was designated VEGF-A. Multiple isoforms of VEGF-A exist due to alternative splicing of the Vegfa gene; however, the 165 amino acid variant in humans (VEGF-A165) is the most potent and prevalent among the major isoforms expressed during pathologic NV.19 The potential roles for VEGF-A signaling in the pathophysiology of CNV have become the focus of numerous translational investigations. CNV membranes surgically excised from patients with AMD were strongly reactive with anti–VEGF-A antibody,20,21 and pharmacological inhibition of VEGF-A decreased experimental laser-induced CNV in animal studies.22,23 These data ushered in an era of rapid development of VEGF-A-targeted therapies for use in patients with neovascular AMD. Further details on VEGF biology and VEGF-targeted therapies will be addressed in subsequent chapters.
Regulation of Angiogenesis During CNV by the Extracellular Matrix
The pathogenesis of CNV requires the invasion of preexisting extracellular matrix (ECM) and the formation of new avenues of vasculature. During this process, specific enzymes are expressed, which degrade the basement membranes allowing for subsequent vascular in-growth. Surgically excised CNV tissue contained fibrin and fibronectin,24 both of which are important in ECM-guided angiogenesis. Integrin receptors, which regulate cell signaling through interaction with various components of the ECM, are also important in CNV formation25 and have been successfully targeted in animal models of CNV.26,27 Several years ago, a link was discovered between proangiogenic factors found in CNV membranes and the upregulation of specific matrix-metalloproteinases (MMP-2 and MMP-9), which are capable of ECM breakdown.28–30 Tissue inhibitors of MMPs (TIMPs), which prevent dissolution of ECM by MMPs, are also expressed in CNV membranes and have provided a novel therapeutic target.31,32 Another proteinase mechanism known to be involved in CNV progression is the urokinase plasminogen activator pathway, which is also being evaluated as an alternative treatment modality.33–35
More novel ECM proteins are being discovered that may be involved in VEGF-A signaling and CNV pathogenesis (Fig. 3.4). The matricellular protein SPARC (secreted protein, acidic and rich in cysteine) is induced by VEGF-A and affects remodeling, cellular migration, and angiogenesis.36,37 SPARC decreased the stimulatory activity of VEGF-A on endothelial cell proliferation,38 but at this time, the exact mechanism of interaction between SPARC and VEGF-A is unknown. Further investigations showed that intravitreous VEGF-A paradoxically suppressed CNV when injected after laser injury, whereas mouse eyes treated with VEGF-A prior to laser injury exhibited the typical proangiogenic response and enhanced CNV.39 This unexpected biphasic VEGF-A activity may be due to decreased SPARC levels during the acute phase of laser-induced injury. These data may dually help improve the scheduling and dosing of VEGF-A targeted therapeutics in order to restrict pharmacologic effects to the intended action of suppressing and reversing CNV. In addition, SPARC expression is restricted to the macula in primates, which may help explain the macular predilection of CNV in humans.40
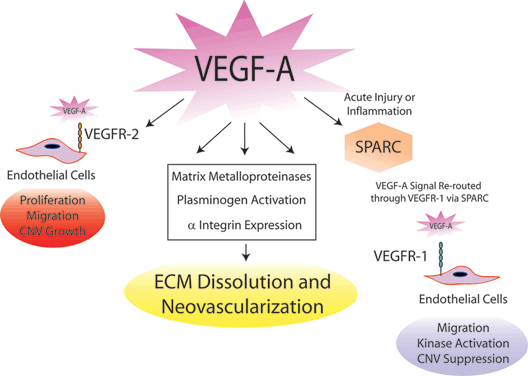
FIGURE 3.4. Modulation of the ECM with injury and VEGF-A upregulation. VEGF-A is a major factor in the pathogenesis of CNV through its direct effects on endothelial cell proliferation and modulation of the ECM. Integrins, plasminogen factors, and MMPs are all upregulated in response to VEGF-A, contributing to ECM degradation that promotes neovascular invasion. VEGF-A is also able to curb the endothelial cell responses through its interaction with SPARC, which reroutes signal transduction away the proangiogenic pathways of VEGFR-2 to the nonproliferative cellular pathways of VEGFR-1.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


