FIGURE 27.1 Fundus photographs showing the typical presentation of XLRS. Left panel: Affected male with spoke–wheel pattern of foveal cysts. Right panel: Affected male with “vitreous veils” of the inferonasal quadrant, retinal pigmented epithelium (RPE) reticular degeneration, and visible inner leaf schisis margin.
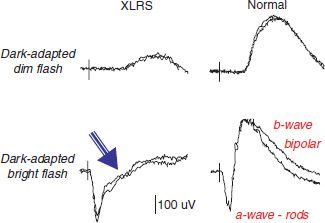
FIGURE 27.2 Scotopic full-field ERG traces from an affected XLRS male (45 years old), harboring a c354del1- ins18 mutation of the retinoschisis (RS1) gene. XLRS affected male shows typical ERG with b-wave reduction but a-wave preservation in dark-adapted recordings, compared with a normal individual. The arrow indicates the characteristic “electronegative” waveform with b-wave smaller than the a-wave.
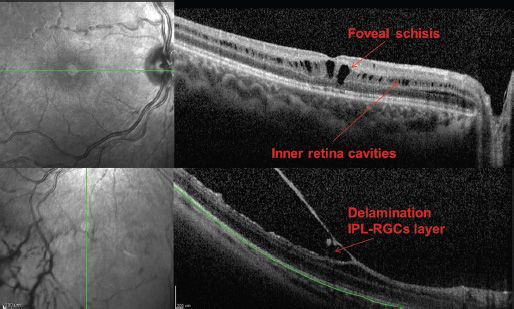
FIGURE 27.3 Infrared images (left side) and OCT (right side) scans from an XLRS boy (12 years old). Top panel: Macular scan, showing foveal schisis and parafoveal inner retina cavities. Lower panel: Peripheral scan, showing the wide delamination of the retina. Images were taken by Spectralis OCT (Heildeberg Engineering).
Environmental and genetic factors, including modifier genes, are predicted to account for some of the phenotypic variation in an XLRS mouse model (8) and, hence, are possible contributors to the wide phenotypic heterogeneity found across the ages of members of XLRS families (9,10). However, modifier genes have not been described for human XLRS.
Contusive trauma can exacerbate vision loss in XLRS patients. Affected males lack a fully functional retinoschisin protein that normally participates in holding retinal layers together and that contributes to the integrity of the retinal architecture though its adhesive properties (11–13). Hence XLRS disease is believed to leave the retina vulnerable to mechanical shock and injury. Prudent medical advice to XLRS males includes avoiding excessive body contact sports. Environmental factors are not currently suspected of contributing to the etiology or progression of the condition.
The RS1 gene for XLRS was identified on the X chromosome by positional cloning in 1997 (14). The RS1 gene contains 6 exons that encode a 224-amino-acid protein called retinoschisin. More than 150 mutations in RS1 have been reported, and a comprehensive list of allelic variants (sequence polymorphisms) is maintained through the Retinoschisis Consortium database (15). Most variants are missense mutations that are clustered in the discoidin domain (exons 4 to 6) and lead to misfolded protein or even functional loss. Founder effects are recognized, as 95% of XLRS patients with Finnish ancestry have one of three mutations in the RS1 gene: p.E72K (c.214G>A), p.G74V (c.221G>T), and p.G109R (c.325G>C) (16). Exons 1, 2, and 3 of the RS1 gene are susceptible to major deletions, splice site mutations, and introduction of a stop codon, all of which would cause a truncated or absolute loss of protein. We observed that less severe mutations tend to occur in exons 4 to 6, and the more severe mutations involve exons 1 to 3 (9,17–19).
XLRS is inherited in classical X-linked recessive manner and is not a syndromic condition. Carrier women have a 50% chance of transmitting the disease-causing mutation in each pregnancy. Males who harbor the mutation are affected, and they transmit the mutation to all their daughters and none of their sons. Females who inherit the mutation will be carriers and nearly always are asymptomatic but can transmit the trait to their sons.
Sporadic cases of bilateral symmetrical maculopathy, without known family history of other affected males, can be particularly challenging to the clinician who is trying to establish a diagnosis and prognosis for the patient and family. Electrophysiologic testing and supplemental molecular genetic analysis are key diagnostic aids. Mutation analysis can identify mutations in the RS1 gene in at least 90% of affected males (20). The value of supplemental molecular genetic analysis is exemplified by the report of a patient with the referring diagnosis of bull’s-eye maculopathy and selective reduction in the b-wave of the ERG (21). Clinical findings suggested the possibility of XLRS, and the patient was subsequently shown to have a novel mutation (p.A100P) in the RS1 gene (21).
The methods to grade clinical severity of XLRS disease are not standardized and vary considerably (3,22–27), which makes it difficult to harmonize definitive genotype–phenotype correlations across different reports. Disease severity and clinical characteristics associated with missense mutations are thought to present with relatively similar and with modest clinical severity (23,27). Our recent reports (18,19,28) indicate that RS1 mutations that disrupt the formation of retinoschisin protein and give a null-protein phenotype, along with mutations that cause loss of a cysteine residue with consequent anomalous protein folding, cause more severe disease with major reduction of b-wave amplitudes and b/a ratios and with delayed a-wave implicit time. By contrast, RS1 mutations that cause conservative amino acid substitutions tend to be less severe. Many examples have been published of severe phenotypes in families with RS1 deletions or splice site mutations that produce premature truncated or no retinoschisin protein; severe disease included retinal detachment and extensive macular lesions, with low visual acuity and subnormal or undetectable b-wave and reduced a-wave amplitudes (1,22,26). By contrast, patients with conservative RS1 missense mutations likely to alter protein structure minimally frequently had normal or measurable ERG b-waves and/or normal retinal periphery and/ or only a blunted macular reflex (22).
Despite these general patterns, gene-to-clinical correlation remains difficult, as even family members with the same mutation or mutation type can exhibit considerable variation (9,10,29,30). Severe XLRS disease, including low vision, vitreous hemorrhage, and varying extent of peripheral schisis, was reported within a Chinese family with a conservative missense mutation (10), and three adult XLRS men with a cysteine residue mutation had mild clinical disease and wellpreserved ERGs (29). Phenotypic variability was found even between identical twin boys presenting with nystagmus in early infancy and similar fundus involvement limited to foveal schisis. Neither twin subsequently experienced severe anatomical complications of the disease. Refraction of one twin at age 2.3 years was +5.00 +2.50 ×105 (OD) and +6.00 +2.25 ×90 (OS). Although his vision remained stable throughout his early childhood, he had only 20/200 acuity when last tested at 22 years of age. His identical twin brother was less hyperopic, at +3.75 OU, and always had less severe nystagmus and has maintained approximately 20/40 acuity in both eyes.
Photoreceptors are an abundant primary site of retinoschisin synthesis (31). Retinoschisin was also found in the inner retina, which suggests that the protein is secreted by or transported to sites beyond the photoreceptors where it is produced. Retinoschisin can be secreted by differentiated retinoblastoma (Weri-Rb1) cells, implying that the protein is produced by photoreceptors and then secreted (31). Antibody to retinoschisin binds to photoreceptors and bipolar cells but not to Müller cells (32), challenging the long-held hypothesis that retinoschisin was a Müller cell defect. We observed that retinoschisin is expressed by nearly all retinal neurons during development and in the adult retina of mice (33).
The retinoschisin protein has a single discoidin domain that is thought to help maintain cell adhesion to extracellular matrix proteins and mediate cell–cell interactions (12,34). This may be particularly important in maintaining the photoreceptor synapse with the bipolar cell (11). Retinoschisin may also help regulate the fluid balance between photoreceptor compartments (35). This hypothesis is based on the co-localization of retinoschisin with the NaK-ATPase, a retinal cationic channel (12,36).
Three mouse models of XLRS have been generated, and all result in deficiency of endogenous murine retinoschisin (11,37,38). The scotopic ERG of the male Rs1- KO mice mimics that of XLRS patients and has selective b-wave reduction. One mouse model also shows an additional severe effect in the cone ERG (11). The retina of the Rs1-KO mouse has disorganized architecture, with areas of schisis in the inner nuclear layer, photoreceptor loss, and shorter outer segments (11,37,39). These findings of the murine XLRS retina support a functional role of retinoschisin in maintaining cell–cell adhesion.
Clinical findings in XLRS vary broadly and can mistakenly be diagnosed as “amblyopia” in young affected boys. Less overt cases of XLRS can be overlooked on cursory fundus examination. Family history indicating an X-linked inheritance of visual abnormality should help in the differential diagnosis.
Affected males have reduced central visual acuity, which typically comes to parental and medical attention during early school-age years, when children manifest difficulties in reading. Vision loss typically is in the range from 20/60 to 20/120, and the eyes frequently are hyperopic with a high level of astigmatism. Fundus examination reveals bilateral foveal schisis in virtually all patients at some point in their clinical histories and can be the only physical finding. The foveal schisis appears as a spoke wheel pattern with areas of retinal folds surrounding the microcysts (Figs. 27.1 and 27.3). The human fovea is incompletely developed at birth and does not reach adult conformation until age 15 months (40). Hence, one can anticipate that the “spoke–wheel” intraretinal parafoveal cysts of XLRS may not be fully clinically apparent at birth.
The maculopathy may progress later in life and develop an atrophic stage of the disease observable by middle age (3). Peripheral retinoschisis involves the inferotemporal region in approximately half of patients, leading to reduced visual field sensitivity (2,23). The schisis may progress toward the central retina or become flat and leave only retinal pigmented epithelium (RPE) abnormalities or retinal scars (Fig. 27.1). Peripheral chorioretinal scars may mimic resolved chorioretinitis. Macular scars can also occur
Optic nerve pallor, dragged disc appearance, or optic nerve head neovascularization (41,42) has been described. Fewer than half the XLRS cases have “vitreous veils” consisting of partial-thickness retinal layers delaminating into the vitreous, along with overlying retinal vessels, which can bleed and cause vitreous hemorrhage. Full-thickness retinal detachment occurs only occasionally, and attempted repair is not successful. The frequency of retinal detachment is <10% in our clinical practices. XLRS patients have an unusual whitish fundus appearance when the dark-adapted retina is exposed to light, termed the Mizuo phenomenon (43,44), believed to result from abnormal potassium processing in the retina. This phenomenon disappears after vitrectomy, presumably as a result of removing the interface between the retina and the vitreous (45).
Other unusual presentations of XLRS include a macular hole (46,47), pseudomacular hole (9), strabismus (48), macular dragging and pigmentary changes (49), macular multiple fine white dots (50), bilateral macular detachment (51), vitreous hemorrhage (52), foveal ectopia (53), and retinal white flecks (54) that may simulate fundus albipunctatus (55).
Visual acuity is nearly always subnormal and can reach 20/200 or less even by the grade-school years. Lesch et al. (3) described no substantial differences of mean best corrected visual acuity of “cystic” (<25 years old) versus “atrophic” (>25 years old) XLRS groups, in agreement with previous reports (25,56). Peripheral visual field is affected by peripheral schisis, which can produce absolute scotomas. Color vision is generally unaffected, and patients do not complain of night blindness.
Young patients with XLRS usually have a normal retinal fluorescein angiogram, but older patients may have macular window defects from RPE changes. Peripheral vascular changes can be noted with angiography, with areas of peripheral retinal nonperfusion and leakage of dye from abnormal retinal capillaries (57) and leakage into vitreous veils (41).
The ERG is one of the most clinically useful tests for XLRS. The dark-adapted ERG shows reduced b-wave amplitude, while the a-wave frequently remains normal (19,58). This ERG configuration is termed an electronegative response because the positive-going b-wave fails to return to above baseline from the negative-going a-wave (Fig. 27.2). Sixty percent of XLRS eyes had the “electronegative waveform” in a cohort of 68 affected men (19). As the b-wave originates from activity of depolarizing bipolar cells that lie postsynaptic to the rods (59–61), this implies primary involvement of the inner retina in XLRS.
Although an electronegative ERG response is not pathognomonic exclusively of XLRS, only a few other retinal dystrophies result in this waveform configuration, and finding this ERG type in a suspect male is clinically confirmatory of XLRS. A normal ERG does not, however, rule out the possibility of XLRS, because patients with a confirmed mutation in the RS1 gene may present with a normal scotopic ERG b-wave (19,20,62) also in the presence of typical fundus findings of XLRS (9). Carriers of XLRS cannot be identified by electrophysiologic analysis, because the a- and b-waves of the ERG are normal. Final thresholds of the dark-adaptation curve and the phases of cone and rod adaptation occur at the appropriate time. The Arden ratio of the electrooculogram is normal (58).
Although some patients with RS1 missense mutations have reduced a-wave amplitudes, the a-wave amplitude in XLRS generally remains normal, which has raised the question whether photoreceptors are affected in XLRS (19), as retinoschisin protein is heavily expressed in photoreceptor inner segments and in the photoreceptor–bipolar cell synapse (32,63).
Alterations in the multifocal ERG (mfERG) have been found with both the XLRS mouse model and in affected men (64,65). Although the relative sensitivity is unknown compared to the full-field ERG, mfERG may be useful to study the severity of macular involvement and to identify some carriers of XLRS. Kim et al. (4) reported mfERG changed in two of nine female obligated carriers of the disease, in normal-appearing fundus.
Optical coherence tomography (OCT) has revealed schisis lesions that frequently lie deeper in the retina than previously thought, often in the midretinal layers (66) (Fig. 27.3). Areas appearing clinically intact can show shallow schisis across the macula, termed flat or lamellar schisis (24) found in the macular area between the arcades and associated with an abnormal ERG.
Gerth et al. (67) showed extrafoveal schisis within the outer and inner nuclear and ganglion cell layer, alone or in combination, and photoreceptor outer and inner segment layer disruption in XLRS patients by OCT. Also Gregori et al. (68) identified cystoid spaces accounting for retinal splitting in the inner nuclear layer and outer plexiform layer. Small cysts, separated from the foveal splitting, can be found in the outer nuclear layer and in the ganglion cell layer and/or nerve fiber layer by OCT.
We find that OCT changes may correlate with surgical outcomes. In one case of a patient who had bilateral surgical intervention, one eye with good recovery of visual function occurred in the eye in which OCT macular schisis resolved on OCT. However, in the fellow eye that failed to recover visual function, the macular schisis remained.
The OCT combined with clinical appearance provides one classification system for XLRS (Table 27.1). Patients were classified into four types based on the presence of foveal cysts only (type 1), foveal cysts by clinical examination plus lamellar macular schisis detected by OCT (type 2), findings of type 2 XLRS plus peripheral schisis (type 3), or foveal cysts and peripheral schisis (type 4) (24). Lesch et al. (3) used OCT in their large cohort of XLRS subjects to understand the progression of disease across lifetime and the effect of age on the phenotype and genotype in XLRS. The authors found correlations between younger age and thicker foveas and between younger age and better b-wave amplitudes. Based on these findings, the authors proposed the definition of a (i) cystic form of XLRS found before 26 years of age or (ii) an atrophic form of XLRS found 26 years of age or older. In the younger group, there were OCT findings of (i) increased foveomacular thickness by cysts and in the older group of patients there was (ii) central thinning from macular atrophy. We have proposed that functional worsening of ERG responses over time be included in determining the progressive nature of XLRS disease (9).
TABLE 27.1
Classification system for XLRS

XLRS, X-linked retinoschisis; OCT, optical coherence tomography.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


