FIGURE 26.1 A: Hematoxylin- and eosin-stained section from an RB specimen. Region of viable tumor cells demonstrating rosette formation. A prominent Flexner-Wintersteiner rosette is indicated (arrow). A tumor vascular complex is present at the top right-hand corner (×25). B: Toluidine blue–stained section from an RB specimen showing fleurette formation, an expression of photoreceptor differentiation (×100).
Cell death usually is very prominent in RB specimens, and extensive areas of cell death are interspersed with zones of viable tumor cells surrounding blood vessels. A number of investigations suggest that tumor cells displaced from feeder vessels by more than 100 µm undergo ischemic necrosis. This observation has been attributed to the metabolic demands of this highly active tumor outstripping the supply of nutrients (61). Although necrotic portions of RB tumors do not characteristically provoke an inflammatory response, massive necrosis in large tumors may occasionally become clinically relevant by producing marked inflammation that involves surrounding tissues and simulates orbital cellulitis (62).
Apoptosis plays a critical role in growth regulation of many tumor types. This form of cell death has been reported in RB primary tumors and in cell lines and has been recognized as the primary mechanism of RB cell death in response to radiation, cytotoxic chemotherapy, and other therapeutic approaches (63,64). Cell death in RB is frequently accompanied by dystrophic calcification. In one clinical series, more than 80% of RB tumors showed calcification, and the degree of calcification appeared to be related to tumor size, with small tumors tending to be less calcified. Extraocular RB tumor does not typically demonstrate calcification (65,66).
Tumor vasculature is another critical histopathologic feature of RB. The development of an intrinsic tumor blood supply, termed tumor angiogenesis, is critical to the growth of primary tumors and also influences metastatic extension by providing access to the systemic vasculature. One significant mechanism promoting angiogenesis in tumors is the production of vascular endothelial growth factor (VEGF). VEGF mRNA and protein have been demonstrated in RB cells, and the expression of VEGF is increased by hypoxia (67–70). Vascular endothelia at intraocular sites distant from the tumor may respond to high intraocular levels of VEGF by increasing expression of VEGF receptors. An increase in VEGF receptors could have relevance for neoplasm-related ocular angiogenesis, such as iris neovascularization, which is occasionally observed in RB patients (69). Leukocytes, particularly cells of the mononuclear phagocyte series, have been observed in RB primary specimens in association with tumor vasculature. The role of these cells in immune surveillance, tumor development, and angiogenesis remains to be determined (56,57).
Left untreated, RB grows relentlessly, filling the ocular cavity and eventually spreading extraocularly. Four primary patterns of intraocular RB growth have been described (54). The endophytic growth pattern is characterized by growth of tumor toward the vitreous space; in this case, the tumor mass may be directly visualized with the ophthalmoscope (Fig. 26.2). In the exophytic growth pattern, tumor grows from the outer retina toward the choroid, elevating the retina and presenting as serous retinal detachment (Fig. 26.2). In practice, most RB patients display a combination of these two growth patterns; this is termed a mixed growth pattern. A fourth pattern, known as diffuse RB, is clinically significant. Although this form is rare, accounting for only 1% to 2% of all RB cases, the tumor insidiously infiltrates the retina, producing plaque-like thickening. The clinical signs in this form are subtle, with consequent delays in diagnosis (71–73).
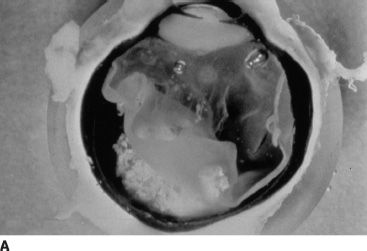
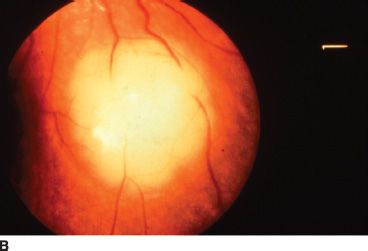
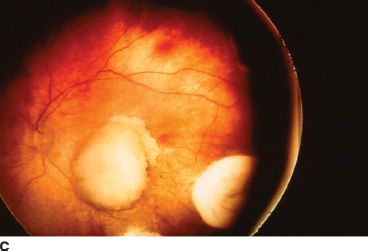
FIGURE 26.2 A: Gross pathology specimen demonstrating the exophytic pattern of RB tumor growth. The chalky white tumor mass at the posterior pole had prominent calcification, arose from the retina, and grew in a scleral direction. Such tumors have a tendency to spread in the subretinal space. B: Clinical ophthalmoscopic appearance of an exophytic RB. The tumor demonstrates a pale yellow color and is covered by the overlying retina, which often has a serous detachment. The retinal vessels overlying the tumor tend to be dilated and tortuous. C: Multiple endophytic tumors in a patient with heritable RB. These tumors appear as pale chalky masses due to prominent calcification.
RB cells are poorly cohesive; they readily disseminate throughout the globe. One mechanism underlying loss of cellular cohesion in RB may include deranged expression of intercellular adhesion molecules such as N-cadherin (74,75). RB cells are shed into the vitreous as small spheroids called vitreous seeds (from endophytic growth) or into the subretinal space (from exophytic lesions). RB tumor cells frequently disseminate away from the main tumor mass, involving ocular structures such as distant retina, lens capsule, or anterior chamber. This dissemination can result in secondary ocular complications such as glaucoma or pseudouveitis (54). Retinal seeding can occasionally mimic multicentric RB, leading to the inaccurate diagnosis of germinal mutation. In large tumors with extensive vitreous and subretinal seeding, the distinction between unifocal and multifocal disease may be virtually impossible to determine clinically.
RB may spread extraocularly and can extend outside the eye by a number of routes. The most frequently observed route of extension is through invasion of the central nervous system (CNS) down the optic nerve, followed by direct extraocular spread and involvement of the orbit. Tumor cells may invade the optic nerve directly or may invade the leptomeninges, gaining access to the orbital apex, optic chiasm, base of the brain, and subarachnoid space (54). Extension of RB cells into the optic nerve is considered a major histopathologic risk factor for the development of metastases, particularly when the tumor extends beyond the lamina cribrosa. If the optic nerve is involved posterior to the line of surgical transection at the time of enucleation, patients demonstrate a dramatically worsened clinical course (76–81). RB tumor cells may also invade the retinal pigment epithelium, Bruch membrane, and choroid. Exposure to the rich choroidal vasculature provides tumor cells access to emissary vascular channels and ultimately to the systemic circulation. The sclera may also be directly invaded by tumor cells. It has been suggested that the exophytic growth pattern is associated with increased risk for choroidal or orbital involvement and metastases, although this hypothesis remains clinically unproved (54,76). It has also been suggested that massive invasion of the choroid is an independent risk factor for disease dissemination (77–84). Tumor cells that spread hematogenously produce widespread metastases to lungs, brain, bone, and other viscera. Lymphatic dissemination of RB may occasionally occur in cases that extend anteriorly and allow tumor cells access to the bulbar conjunctiva and to the eyelid lymphatics (54).
The complex molecular interactions among tumor cells, parenchymal tissue, and extracellular matrix are currently not fully understood. Recent research suggests that a growth and invasion advantage may be conferred on subgroups of tumor cells that become independent of normal mechanisms for cell death or apoptosis and avoid tissue constraints, allowing spread to distant sites. These clones of more aggressive cells arise through ongoing mutagenic and epigenetic phenomena within the tumor. Some molecular mechanisms identified in RB and other tumors that could facilitate these changes include up-regulation of telomerase activity, N-myc oncogene amplification, p53 inactivation, changes in cell adhesion molecules (e.g., cadherins), altered expression of integrin subunits by tumor cells, and increased expression of angiogenic factors (85–88). Greater understanding of these critical molecular processes is likely to yield better and more specific therapies for this disease in future.
OVERVIEW OF RETINOBLASTOMA GENETICS
Retinoblastoma Gene and Protein Product
RB is one of very few cancers in which a single genetic mutation predicts disease development at very high penetrance. The presence of an underlying genetic mutation in RB was predicted by Knudson in 1971, and this prediction led to identification of the RB susceptibility gene locus, with subsequent cloning of this tumor suppressor gene in 1987. Identification of the RB1 gene provided definitive evidence that loss of tumor suppressor gene function is important in the etiology of cancer. Although it has recently been proposed that other genes may be involved in the development of RB, in order for RB to arise, a mutation on both alleles of the RB1 gene is required (15).
The RB1 gene encompasses 180,000 base pairs on chromosome 13q14 and contains 27 exons. The gene encodes a 4.7-kDa message that results in a 105-kDa nuclear phosphoprotein. The RB protein, known as pRB, is a transcriptional modulator expressed throughout development in all cells of adult humans (89,90). The protein has two conserved regions where important functional domains reside and which also represent sites for viral oncogene binding (e.g., SV40 T-antigen, adenovirus E1A, and HPV E7). Oncogenic viruses are capable of inactivating pRB through binding, thereby inducing malignant transformation. Many of the clinically relevant mutations that have been observed in RB1 affect these conserved regions of the protein (34,91–93). Mutations have also been observed in the promoter region of the gene; these mutations affect recognition sequences for transcription factors (e.g., ATF and SP-1) (94).
Mutations affecting the RB1 locus appear to be very heterogeneous. Few preferential sites for mutation, or “hot spots,” have been identified (34,95). Only 3% to 5% of constitutional mutations in RB1 are cytogenetically recognizable; these include interstitial deletions or translocations involving 13q. The remainder of mutations are submicroscopic, including point mutations, microdeletions, and duplications. In more than 90% of RB1 mutations, deoxyribonucleic acid (DNA) alterations result in a truncated mRNA product (34,89). The second mutation that occurs in the retinal cell of origin of the tumor is a result of chromosomal abnormalities, such as nondisjunction, loss of heterozygosity due to mitotic recombination, or large deletion (89,96). This second event may be more influenced by environmental factors, including ionizing radiation. Therapeutic radiation exposure influences the distribution of nonocular tumors.
The RB protein has a critical and complex role in the regulation of cellular growth and differentiation. pRB binds to more than 30 separate cellular proteins, including transcription factors, nuclear matrix proteins, growth regulators, protein phosphatases, and protein kinases (97). One of the best understood functions of pRB involves regulation of the cell cycle through inhibition of the transcription factor E2F (98,99). In this role, the active hypophosphorylated form of pRB acts at a restriction point in late G1 of the cell cycle to inhibit the E2F family of transcription factors. These factors in turn regulate expression of a large set of genes, including, among others, c-myc, b-myb, cdc2, E2F-1, and the dihydrofolate reductase gene associated with cell division in S phase (98,99). The phosphorylation and dephosphorylation of pRB itself are controlled by a complex series of cell cycle–dependent and cell cycle–independent enzymes, including cyclin–cyclin-dependent kinase complexes (cyclin–cdk) and cyclin-dependent kinase inhibitors (CDKIs) (100,101) (Fig. 26.3).
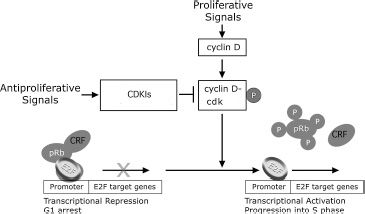
FIGURE 26.3 Schematic representation of pRB function in the regulation of the cell cycle. Active hypophosphorylated pRB blocks cell cycle progression by repressing E2F target genes through binding promoter bound E2F and by recruiting chromatin remodeling factors (CRFs) including histone deacetylase. Phosphorylation of pRB is controlled by cyclin–cyclin-dependent kinase (cyclin–cdk) complexes resulting in inactivation of pRB, allowing transcriptional activation of E2F target genes. Cyclin-dependent kinase inhibitors (CDKIs) positively regulate pRB by inhibiting the negative regulators, the cyclin–cdk complexes.
In addition to its role in cell cycle regulation, pRB has important effects on cellular differentiation. These effects include cell cycle arrest, suppression of apoptosis, and transcriptional activation of tissue-specific genes (90). These effects may be particularly important in tissues such as muscle, bone, fat, and nerve (102–106).
Despite the ubiquitous expression of pRB in all dividing cells, germline mutations in humans appear very specifically to cause RB and tumors of mesenchymal origin. These mesenchymal tumors include osteosarcoma, leiomyosarcoma, and malignant fibrous histiocytoma (107). In contrast, somatic alterations in pRB, which occur mainly through functional inactivation of upstream regulators, are involved in the progression of many common cancers. These include carcinoma of the breast, bladder, and prostate; small cell lung cancer; leukemia; and glioblastoma (100). For example, mutation of p16Ink4a (a CDKI) and amplification of cyclin D, both upstream regulators of pRB, are frequently observed in these malignancies (108,109). The vulnerability of RB1-deficient cells in the retina and certain connective tissues to tumor development in the presence of RB1 germline mutation has never been adequately explained. It is possible that this vulnerability arises from a critical requirement for pRB in terminal differentiation of these tissues.
Although mutation in both RB1 alleles initiates RB, other chromosomal abnormalities and mutations have also been consistently observed. These other consistent genetic abnormalities include trisomy 1p, loss of heterozygosity for chromosome 17, and isochromosome formation at 6p (110–112). These abnormalities could represent secondary changes due to genetic instability and could play a role in mediating malignant progression, allowing clones of malignant cells to acquire selective growth advantage. Interestingly, reconstitution of RB cells, RB-null carcinoma cells, or sarcoma cells with functional pRB only partially modulates the malignant phenotype (113). This suggests that other consistent genetic alterations may be important for development of the full tumor phenotype.
Clinical Genetics
Heritable Retinoblastoma
Although only 10% of RB patients have a family history of this disease, new germline mutations account for an additional 30%, meaning that 40% of RB patients have a heritable form of RB. Patients with heritable disease have a mutation (or first “hit,” according to Knudson’s hypothesis) involving RB1 in every cell of their bodies. This is termed a germline mutation because the mutation either was inherited from a parent or, more commonly, developed at the one-cell stage of embryonic development in the affected individual. Individuals with germline mutations in RB1 are capable of passing on the mutation to their offspring. Mutation in the other copy of RB1 (the second “hit” described by Knudson) occurs in an undifferentiated embryonic retinal cell that gives rise to the malignant tumor. In this heritable form of RB, patients demonstrate bilateral, multifocal retinal tumors, which are pathognomonic for heritable disease. Although the RB gene at the cellular level is recessive (i.e., both RB alleles must be abnormal in order for the tumor phenotype to be expressed) at the level of the individual, the mutation behaves in an autosomal dominant manner, with 80% to 90% penetrance (114) (Table 26.1).
TABLE 26.1
Clinical and genetic forms of RB
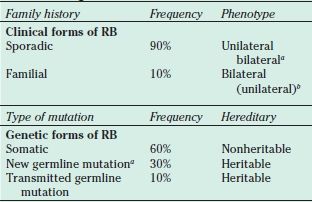
aSome of these cases may represent parental mosaicism.
bFifteen percent of total germline mutations present as unilateral diseases.
RB, retinoblastoma
Nonheritable Retinoblastoma
The remaining 60% of RB cases represent nonheritable or somatic disease. In this nonheritable form, mutations in both RB1 alleles (i.e., both the first and the second “hits”) occur within a single retinal cell of origin for the tumor. Such events are rare; therefore, these patients demonstrate a single unilateral, unifocal tumor. Unilateral disease presents, on average, at a later age than does bilateral disease.
It should be recognized that approximately 15% of patients who present with unilateral RB have the heritable form of this disease, but by chance they have developed a tumor in only one eye (115–117). The relationship between the clinical and the genetic forms of RB is summarized in Table 26.1.
Second Tumor Predisposition
As a consequence of germline mutation in RB1, patients with heritable RB should be regarded as having a genetic cancer predisposition syndrome. The clinical consequences of this syndrome are that these patients develop not only multiple, bilateral retinal tumors, but they also demonstrate a severalfold increased risk for developing second malignancies in later life, including osteogenic sarcoma, fibrosarcoma, and melanoma (34,35). These second nonocular tumors may occur anywhere throughout the body, although their distribution appears to be increased within the radiation field following radiotherapy (34,35,37,118,119). By age 40 years, 30% of heritable RB patients will have developed a second nonocular cancer (37). Additionally, approximately 8% of hereditary RB patients will develop midline intracranial primitive neuroectodermal tumors (PNETs, formerly called trilateral RB), which frequently occur in the pineal region (36). These tumors usually occur in conjunction with bilateral disease, although PNETs have also been described in patients with unilateral retinal tumors (120).
Genetic Mosaicism
It has been recognized that 10% of RB families demonstrate germline or somatic mosaicism (121,122). Mosaicism refers to the presence of two or more cell lineages differing in genotype in an individual. If a mutation develops at the RB1 locus during later stages of embryonic development, the lineage of cells derived from this original mutation-bearing cell will carry an identical RB1 mutation. In contrast, cells derived from unaffected lineages will carry normal copies of RB1. If the mutant RB1 is carried in the gametes but not in the eye, the affected individual could pass the mutation to offspring without personally demonstrating clinical evidence of RB. The practical significance of mosaicism in one of a proband’s parents is that siblings of this proband would be at higher risk for developing the disease than previously believed (123). Therefore, siblings should receive early and regular screening for development of this disease (123). Parents who are mosaic for mutant RB1 may also be at personal risk to develop malignancies in cell lineages that carry the mutant gene. These individuals should receive prompt referral to an oncologist if systemic complaints suggestive of malignancy develop.
Low-Penetrance Retinoblastoma
Forms of RB have been described in families where a proportion of RB1 mutation carriers do not develop eye tumors. This clinical circumstance is termed low-penetrance RB. In other families, RB1 mutation carriers either develop unilateral disease or demonstrate a benign variant of RB called retinoma. This clinical circumstance is described as low-expressivity RB. Low-penetrance and low-expressivity forms of RB have been associated with RB1 mutations that either partially inactivate the protein or reduce protein expression (124,125). Recognition of these kindreds is important because they allow more accurate genetic counseling for affected families (96,114,126).
Current Status of Genetic Counseling and DNA Testing
It is important that all ophthalmologists who treat patients with RB inform patients and families of the potential heritable nature of this disease. Families also should be made aware that germline mutation in RB1 confers a lifetime second tumor predisposition. Referral to a pediatric oncologist for second tumor surveillance is indicated.
Genetic counseling is often performed by the ophthalmologist in conjunction with a clinical genetics service (127–129). A careful and complete family history should be taken at the time of the initial referral. Examination of relatives for retinoma or regressed RB can be very helpful in some cases, as this suggests carrier status for the affected relative.
CLINICAL PRESENTATION AND DIFFERENTIAL DIAGNOSIS
When there is a family history of RB, most cases in the United States are detected as part of RB screening programs. In these siblings or offspring of RB patients, the diagnosis usually is made at a very early stage in infancy when the tumors are small, offering the best opportunity for treatment aimed at preserving the eye and the vision.
In unaffected families, RB presents either incidentally or following referral by a family physician or pediatrician. Often the first sign of the disease is a white pupil (leukocoria) or strabismus, usually noticed initially by a patient’s parent or relative. White pupil, or leukocoria, was first described as a presentation for RB by Hayes in 1767 and unfortunately remains the most common presenting sign of this disease today (in ~60% of cases) (Fig. 26.4) (130–133). Strabismus is the second most frequent presentation of RB (in ~20% of cases), and this observation reinforces the need to perform a dilated retinal examination in all new patients who present with strabismus. Other less frequent presentations for RB include intraocular inflammation, iris neovascularization, hyphema, pseudohypopyon, preseptal/orbital cellulitis, and glaucoma (Fig. 26.4). With increased awareness and the advent of early screening programs, some patients may be found at a very early stage with no signs apart from the presence of a small retinal lesion.
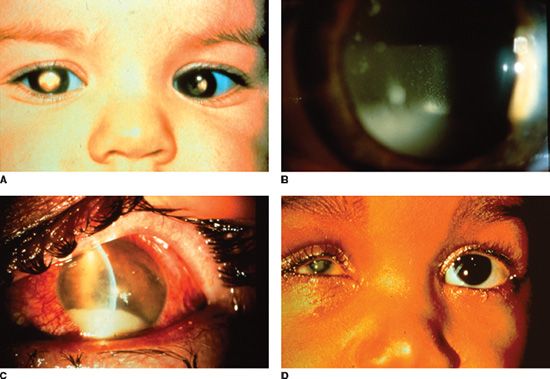
FIGURE 26.4 A: Heritable RB patient presenting with bilateral leukocoria, the most commonly reported presenting sign of this condition. B: RB cells have a propensity to detach from the primary tumor mass and spread throughout the eye, as illustrated by a patient with endophytic RB who developed marked tumor cells in the vitreous, resembling a vitritis. C: Occasionally tumor necrosis may produce an inflammatory response associated with a red eye and conjunctival injection as seen in this patient. The patient also has a tumor hypopyon. D: Massive tumor necrosis in a phthisical eye of a patient with RB has resulted in marked inflammation involving the orbit, simulating an orbital cellulitis with lid swelling and pain on eye movement.
The clinical presentations of RB that simulate inflammation are associated with the greatest diagnostic difficulty. True inflammation may arise in an eye with RB due to extensive tumor cell necrosis. This situation frequently occurs with larger tumors, and the accompanying inflammation may simulate preseptal or orbital cellulitis. If RB is suspected, identifying the apparent cellulitis as a masquerade syndrome is often straightforward. Children with true orbital cellulitis exhibit fever, leukocytosis, and sinusitis. These findings are generally absent in patients with RB. The presence of normal sinuses and evidence for an intraocular mass are easily documented with computed tomography (CT) imaging (Fig. 26.4).
Another less straightforward clinical presentation for RB involves friable tumor cells that detach from the main tumor mass and subsequently spread throughout the eye. These tumor cells may simulate the white blood cells of a hypopyon and may suggest uveitis as an initial diagnosis (Fig. 26.4). Secondary glaucoma, due to mass effect from the tumor producing angle closure or due to secondary iris neovascularization, occasionally may produce a diagnostic challenge.
Rarely in the developed world, patients may present with evidence of extraocular spread, metastatic disease, or CNS symptoms due to a PNET. Proptosis, although rare as a presenting sign of RB in industrialized societies, unfortunately is a common presentation in less-developed countries. Proptosis was the most common presentation of RB (65% of cases) in one study from India (33). This presentation carries a poor prognosis because it is frequently associated with advanced disease and extraocular spread into the orbit.
RB occasionally may occur in association with other congenital abnormalities, including cleft palate, cardiovascular defects, infantile cortical hyperostosis, dentinogenesis imperfecta, incontinentia pigmenti, or familial congenital cataracts. Patients also may demonstrate features of 13q deletion syndrome, associated with deletions in the distal part of chromosome 13q. These patients demonstrate mental retardation, broad nasal bridge, hypertelorism, microphthalmia, micrognathia, cleft palate, or foot and toe abnormalities (134,135).
Clinically, an endophytic tumor appears as a white- or cream-colored mass that penetrates the internal limiting membrane of the retina and demonstrates prominent white chalky areas that represent intrinsic calcification. The vessels associated with endophytic RB usually are small, irregular tumor vessels that stand in marked contrast to the dilated or aneurysmal blood vessels observed in Coats disease. Endophytic RB may be associated with seeding of tumor cells into the vitreous space. These clumps of cells present as snowball-like spheroids within the vitreous cavity with intervening clear spaces. Occasionally, vitreous seeding from RB may be so extensive that the anterior chamber is involved. This presentation with a tumor hypopyon can resemble endophthalmitis (Fig. 26.4).
In its exophytic form, RB tumors usually are yellow to white in coloration and are observed deep to the overlying retina, which is often serously detached. The retinal vessels overlying the tumor may be dilated and tortuous. In contrast to serous detachments from other causes, an underlying mass with characteristic features usually can be demonstrated by ultrasound, CT, or magnetic resonance imaging (MRI). Occasionally, intraretinal and subretinal exudation may mimic Coats disease, making diagnosis difficult (Fig. 26.2) (136).
Detached tumor cells in the vitreous or subretinal space have the capacity to implant and to grow at other retinal sites, producing multiple tumor foci. Most large tumors have both exophytic and endophytic features. Very small RBs may appear as pale grayish masses confined to the retina.
The diffuse form of RB is the most difficult form to accurately diagnose. Diffuse RB often presents in older children as a resistant uveitis with mild conjunctival hyperemia and pseudohypopyon. Ophthalmoscopy may reveal a diffuse gray-white opacification and retinal thickening with vitreous seeds. Frequently this form of RB is situated in a more anterior location, underscoring the importance of dilated fundus examination with scleral indentation. In the absence of a well-defined mass with a lower incidence of intrinsic calcification, ultrasound, CT, and MRI may be less informative in establishing the diagnosis of diffuse RB (71–73).
The clinical diagnosis of RB always requires that this malignant tumor be distinguished from other simulating disease processes. In a study of 500 patients referred to the Wills Eye Hospital for suspected RB, only 58% of referred patients were found to have RB on clinical evaluation. The remaining 212 patients received diagnoses of 23 different forms of pseudoretinoblastoma (133,136). The most common simulating lesions were persistent hyperplastic primary vitreous (PHPV) (28%), Coats disease (16%), and presumed ocular toxocariasis (16%), followed by retinopathy of prematurity and retinal hamartoma. Other simulating lesions included hereditary conditions, such as familial exudative vitreoretinopathy, congenital retinoschisis, Norrie disease, and incontinentia pigmenti. Developmental conditions that were mistaken for RB included retinal or optic nerve coloboma, retinal dysplasia, congenital retinal folds, and myelinated nerve fibers. Inflammatory or infectious conditions, including congenital toxoplasmosis or toxocariasis, or cytomegalovirus or herpesvirus retinitis, also can simulate RB. Tumors such as medulloepithelioma, choroidal hemangioma, glioneuroma, capillary hemangioma, and ocular infiltrates from leukemia occasionally can present initially as RB. It should always be recalled in the setting of media opacities, such as hyphema, cataract, or vitreous hemorrhage, that the diagnosis of RB is not excluded until the retina has been thoroughly examined or imaged to exclude a concomitant tumor. RB coexisting with other conditions, such as PHPV or cataract, has been reported (137). In practice, most clinical cases of RB can be diagnosed by careful clinical evaluation by an experienced examiner and the judicious application of supplemental investigations. Table 26.2 summarizes the salient diagnostic features of the more common lesions that simulate RB.
Whenever RB is suspected, a detailed medical history should be taken at initial examination, including a prenatal and birth history. Historical features suggesting a pseudoretinoblastoma should be specifically investigated (Table 26.2). A family history should be obtained, and a detailed family tree should be diagrammed in the medical record. A history of RB or other forms of cancer, as well as a history of ocular malformations, blindness, or enucleation in other family members, should be questioned. Enucleations occurring within a family are often better remembered than the precise diagnosis of RB. The family tree should document the ages and health status of all siblings and near relatives.
TABLE 26.2
Diagnostic features of most common lesions simulating RB
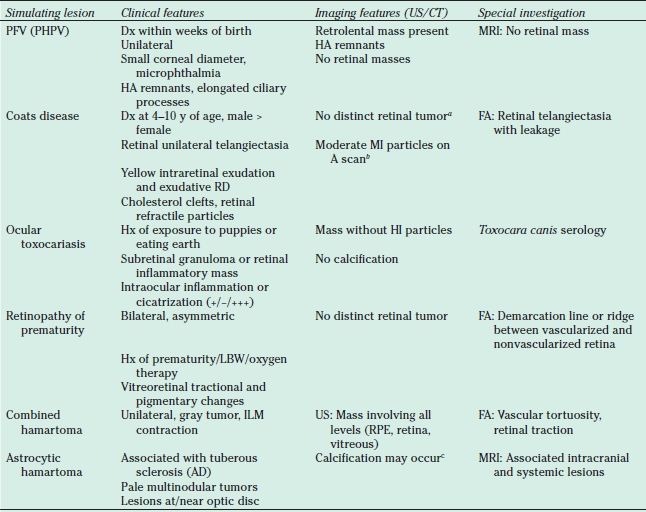
aRB can produce a Coats-like reaction with a large amount of retinal exudation. In these eyes, US can usually detect the retinal tumor beneath the exudate.
bCorrespond to the refractile cholesterol particles seen clinically. These particles are much less reflective than the calcium particles seen with RB (From Shields JA, Shields CL, Parsons HM. Differential diagnosis of retinoblastoma. Retina 1991;11:232–243).
cCalcification is not specific to RB, although it is very characteristic. The calcification in astrocytomas appears clinically as refractile yellow areas vs. the chalky white appearance in RB.
RB, retinoblastoma; PHPV (PFV), persistent hyperplastic primary vitreous (persistent fetal vasculature); CT, computed tomography imaging; Dx, diagnosis; FA, fluorescein angiography; HA, hyaloid artery; HI, high intensity; Hx, history; ILM, internal limiting membrane; LBW, low birth weight; MI, medium intensity; MRI, magnetic resonance imaging; RD, retinal detachment; RPE, retinal pigment epithelium; US, ultrasound imaging; AD, autosomal dominant.
In every child with suspected RB, a complete physical examination is required. Specific features associated with simulating lesions and/or RB syndromes or systemic metastases should be evaluated. For example, the child should be examined for evidence of tuberous sclerosis (e.g., ash leaf spots) or dysmorphic features suggesting a 13q deletion syndrome. Evidence for underlying systemic disease or metastases, such as cachexia, wasting, or developmental delay, should be investigated. Clinical features of PNETs associated with trilateral RB include abnormal eye movements, vomiting, seizure, headache, and changes in cognition. We routinely refer all patients with suspected RB to a knowledgeable pediatric oncologist for a complete history and physical examination.
The ophthalmologic evaluation of a child with suspected RB should be detailed and complete. Visual acuity and visual function should be assessed with each eye patched. The visual potential of each eye is an important determinant for future therapy. External examination of the patient should evaluate for dysmorphic features, lymphadenopathy, or proptosis. Pupils and eye movements should be evaluated for evidence of a concomitant intracranial disease process. Careful anterior segment examination is necessary to evaluate the size of the corneas and the clinical appearance of anterior segment structures. For example, a unilateral small corneal diameter may suggest PHPV rather than RB. Signs of intraocular inflammation should be sought on slitlamp evaluation. Although RB may rarely produce a pseudouveitis or tumor hypopyon, signs of intraocular inflammation are more often associated with granulomatous uveitis, such as toxocariasis, or with other pediatric uveitis syndromes. Intraocular pressures for each eye should be recorded, and the cause of any pressure elevation should be investigated.
Bilateral dilated fundus examination with scleral depression is performed under general anesthesia. Scleral indentation must be performed for 360 degrees of the globe in order to inspect the entire surface of the retina to detect anteriorly located tumors (138). In one series, RB in the periphery of the fundus was detected using scleral indentation in 65% of cases (138). Color fundus photos with a wide-angle contact camera and retinal drawings should be performed to document the size and location of all retinal tumors. This documentation is essential to plan treatment and to monitor subsequent treatment response.
Supplementary investigations are frequently used to aid in the diagnosis and staging of patients referred with RB. Ultrasound is a useful tool, particularly for confirming the initial diagnosis and for evaluating features of intraocular disease. In larger tumors, A-scan echography typically demonstrates high-intensity echoes within the tumor that correspond to areas of tumor calcification. The tumor surface nearest the sclera may be anechoic. B-scan often demonstrates an intraocular mass with scattered highly reflective echoes within the tumor and with attenuation of normal soft tissue signals posterior to the tumor. In patients with advanced disease, exudative retinal detachment, vitreous seeding, and subretinal seeding also may be demonstrated by ultrasound (Fig. 26.5) (139).
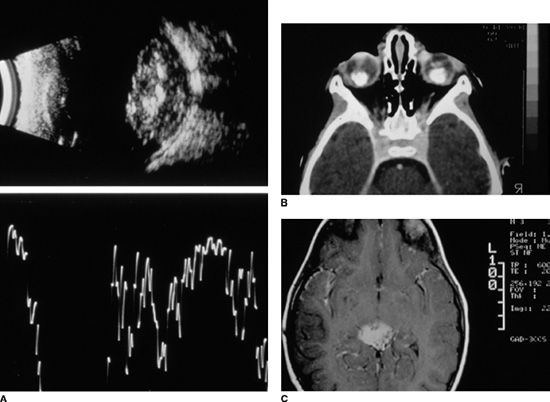
FIGURE 26.5 A: Ultrasound scan of RB showing a retinal mass located at the posterior pole. Both (A) and (B) scans demonstrate that the mass has areas of high internal reflectivity due to intrinsic calcification. Intrinsic tumor calcium may produce a shadowing of the soft tissues behind the tumor. B: CT scanning performed with contrast demonstrates bilateral RB tumors involving the posterior poles with dense calcium deposits. C: T1-weighted MRI scan with gadolinium enhancement demonstrating an enhancing PNET in a patient with heritable RB.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


