FIGURE 25.1 Schematic diagram of a photoreceptor and RPE cells showing many of the cellular pathways and processes associated with retinal degenerative diseases in these cells. Stgd1MD, autosomal recessive Stargardt macular degeneration; LRAT, lecithin retinol acyltransferase; LCA, Leber congenital amaurosis; RDH, retinol dehydrogenase; IRBP, interphotoreceptor retinoid-binding protein; RP, retinitis pigmentosa; RGR, RPE-retinal G protein–coupled receptor 4; RBP4, retinol-binding protein 4; CSNB, congenital stationary night blindness; GNAT1, rod transducin; PDEA/B, phosphodiesterase α- and β GRK1, G protein–coupled receptor kinase 1; CNGA1/B1, rod cyclic nucleotide-gated channel α and β NCKX1, rod Na/Ca–K exchanger; GC1, guanylate cyclase 1; CRD, cone–rod dystrophy; GCAP1/2, guanylate cyclase–activating proteins 1 and 2; CD, cone dystrophy; CNGA3/B3, cone cyclic nucleotide-gated channel; ACHM, achromatopsia; PDE6C, cone phosphodiesterase 6C; MD, macular dystrophy; digRP, digenic RP; RCD, rod–cone dystrophy; USH, Usher syndrome; BBS, Bardet-Biedl syndrome; CHM, choroideremia; ESC, enhanced S-cone syndrome; SFD, Sorsby fundus dystrophy.
In some cases, genes associated with retinal diseases are expressed in other sensory cells as well as the retina. Usher syndrome, characterized by RP and deafness, is typically caused by mutations in genes encoding proteins that are crucial for the development, function, and survival of photoreceptors and hair cells of the inner ear. In some instances, defects in more widely expressed genes can result in a pleiotropic disorder. BBS is an example in which mutations in genes crucial for basal body and ciliary biogenesis, structure, and function cause not only RP but also obesity, polydactyly, diabetes, mental and growth retardation, and auditory deficiencies among other problems. Finally, some genes are ubiquitously expressed in virtually all cells but loss-of-function mutations are only evident in the retina. This is exemplified by ubiquitously expressed RNA splicing genes, such as PRPF3, PRP31, and PRPC8, mutations that cause only RP. Likewise, mutations in the gene for RP2, a ubiquitously expressed protein associated with vesicle trafficking, cause a subset of X-linked RP.
The types of mutations that cause inherited retinal degenerative diseases are also diverse. Missense mutations resulting in single amino acid substitutions are often the most common disease-associated defect. However, other types of mutations including nonsense mutations, splice site mutations, deletions, duplications, and insertions are widely found. Frameshift and nonsense mutations are predicted to produce truncated proteins, which in many cases are unstable and rapidly degraded, thereby effectively resulting in a null allele. In some instances, however, the mutated protein may be highly expressed and have a negative effect, that is, a loss of function, on the wild-type protein as in the case of autosomal dominant diseases caused by mutations in peripherin/rds, rhodopsin, and ELOVL4.
Many studies have been directed toward correlating genotypes with phenotypes for specific retinal degenerative diseases (6,10–12). This, for the most part, has been a daunting task as the phenotype for many diseases results not only from the specific mutation but also other socioenvironmental and genetic factors.
PROTEIN ANALYSIS: GENERAL CONSIDERATIONS
An essential next step in understanding retinal diseases is characterizing the structural and functional properties of proteins encoded by disease-associated genes, defining their roles in cell processes, and determining how disease-causing mutations affect protein structure and function and lead to cell death. Proteins encoded by disease-associated genes participate in a wide variety of cellular processes (Fig 25.1). Virtually all the proteins that function in phototransduction in rod and cone photoreceptor cells are associated with various diseases (Fig. 25.2A). These include rhodopsin (RP, congenital stationary night blindness [CSNB]), rod transducin (CSNB), rod phosphodiesterases 6A and 6B (RP), guanylate cyclase1 (LCA, CRD), guanylate cyclase–activating proteins 1 and 2 (cone dystrophy [CD], CRD), rod cGMP-gated channels A1 and B1 (RP), arrestin (RP, Oguchi disease), rhodopsin kinase (Oguchi disease), rod Na/K–Ca exchanger (CSNB), long and middle wavelength cone opsins (CD), cone cGMP-gated channels A3 and B3 (achromatopsia), cone phosphodiesterase 6C (achromatopsia), and cone transducin (achromatopsia), among others (Fig. 25.1). Enzymes that function in the visual cycle and retinoid processing are also associated with a variety of retinal diseases (Fig. 25.2B). These include ABCA4 (Stargardt disease, CRD, RP), lecithin retinol acyltransferase (LRAT) (LCA14), RPE65 (LCA2, RP), retinol dehydrogenase 12 (RDH12) (LCA13, RP), RDH5 (fundus albipunctatus), interphotoreceptor retinoid-binding protein (RP), and retinal G protein–coupled receptor (RP), among others. In addition, proteins involved in a wide variety of other cell processes are also known to cause retinal degenerative diseases when mutated. Such processes include photoreceptor outer segment morphogenesis, renewal, and structure (peripherin/rds, rom-1, prominin-1, MERTK); protein chaperones (AIPL1); basal body and ciliary structure and transport (CEP290, LCA5, RPGRIP1, RPGR, RP1, USH2A, USH2C, USH2D, BBS1-7); protein and vesicle trafficking (TULP1, RD3, RP2, ARL6); RNA splicing (PRPF8, PRPF31, PRP3, PAP1); lipid biosynthesis and metabolism (ELOVL4, CERKL); ion channels and synaptic structure and transmission (bestrophin1, CACNA1F, CACNA2D4, HRG4, KCNV2, clarin-1); nucleotide metabolism (IMPDH1); extracellular proteins (TIMP3, retinoschisin, fibulin-5, EFEMP1); molecular scaffold (CRB1); retinal development (CRX, NRL, NR2E3); cell adhesion (CDH23, PCDH15); and others.
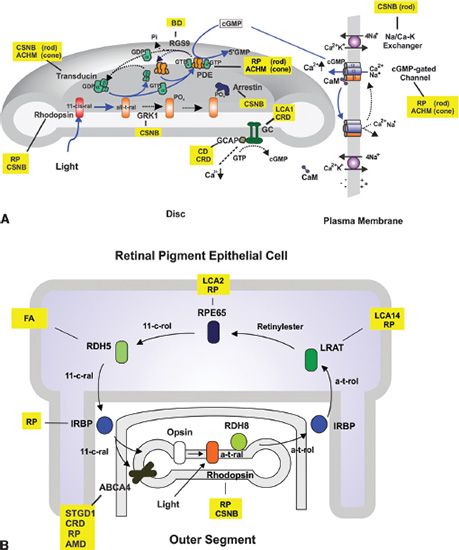
FIGURE 25.2 Diagrams showing phototransduction and the visual cycle and diseases associated with key proteins of these pathways. A: Phototransduction. Light isomerizes 11-cis retinal (11-cis-ral) to all-trans retinal (all-t-ral) within rhodopsin. Activated rhodopsin catalyzes the exchange of GDP for GTP on transducin α-subunit. The transducin α-subunit dissociates from the β- and γ-subunits and activates phosphodiesterase (PDE), which catalyzes the hydrolysis of cGMP. As the cGMP level drops, the cGMP-gated channel closes resulting in a hyperpolarization of the cell. The cell returns to its dark state through (i) the inactivation of rhodopsin via phosphorylation by rhodopsin kinase (GRK1) and the binding of arrestin; (ii) the inactivation of PDE when GTP bound to the transducin α-subunit is hydrolyzed to GDP, a reaction catalyzed by RGS9; (iii) the increase of cGMP levels as guanylate cyclase (GC) is activated by the calcium-binding protein GCAP and low Ca2+ concentration. The latter is caused by the closure of the cGMP-gated channel and continued extrusion of Ca2+ by the Na/Ca–K exchanger and (iv) the reopening of cGMP-gated channel with increased levels of cGMP resulting in the return of the cell to its depolarized state. Solid line (blue) shows photoexcitation reactions; dotted line (black) shows photorecovery reactions. B: Visual cycle showing the key retinoids and enzymes involved in the resynthesis of 11-cis-retinal (11-cis-ral) from all-trans-retinal (all-t-ral) following photobleaching of rhodopsin. All-trans-retinal is reduced to all-trans-retinol (all-t-rol) by RDH8, converted to retinylesters by LRAT, isomerized to 11-cis-retinol (11-cis-rol) by RPE65, and oxidized to 11-cis-retinal by RDH5. The 11-cis-retinal recombines with opsin to regenerate rhodopsin. ABCA4 acting as a transporter plays an important role in the removal of excess 11-cis-retinal and all-trans retinal. Diseases associated with various proteins: RP, retinitis pigmentosa; CSNB, congenital stationary night blindness; CD, cone dystrophy; CRD, cone–rod dystrophy; ACHM, achromatopsia; FA, fundus albipunctatus; LCA, Leber congenital amaurosis; BD, bradyopsia.
In some instances, extensive studies have defined the role of these proteins in photoreceptor or RPE cell development, structure, and function, as exemplified by rhodopsin. However, in most cases disease-associated proteins have been poorly characterized and their roles in cell processes have been surmised based on the presence of structural domains, localization within the cells, and phenotypes of animal models harboring defective genes. Detailed analysis of these proteins at a biochemical level should provide more insight into the role of these proteins in retinal cell biology and diseases but remains a challenge for many research groups.
An understanding of the role of proteins encoded by specific disease-associated genes has been facilitated through analysis of animal models and cell culture techniques (13–17). In most cases, animal models such as mice or zebra fish have been generated to evaluate the effect of gene deficiency on the retina. Complete analysis using a combination of techniques including histology, optical coherence tomography (OCT), electroretinography (ERG), immunocytochemistry, biochemistry, and RNA/protein expression has yielded important clues. In many cases, the characteristic features observed in these animal models compare favorably to those of the human disease and hence serve as valuable tools for detailed analysis of the role of the proteins in retinal cell biology and physiology and further serve as models for the development of therapeutic treatments for specific diseases (14,16–18). However, in some instances the phenotype of the knockout mice does not correspond well with the phenotype observed in humans. For example, many mouse models for Usher syndrome show little if any retinal degeneration compared to patients with Usher syndrome, although they typically exhibit a deficiency in hearing (19). This may result from differences in the retinal structure and physiology between mice and humans or alternatively it may reflect the fact that humans live longer and are exposed to more intense light conditions. Likewise, a knockout mouse may not represent a useful model for some diseases including many autosomal dominant inherited diseases. In these cases, it may be necessary to develop transgenic mouse models in which the mutated gene is expressed along with the normal gene or in which the mutated gene replaces the normal gene to more fully evaluate the effect of the mutation on retinal structure and function (18,20).
In addition to animal models, information on the role of proteins in retinal structure and function has come from analysis of interacting proteins and protein networks (21–23). Proteomic and bioinformatic approaches have been successfully applied to the analysis of proteins associated with LCA, BBS, and Usher syndrome and in some cases have defined plausible networks of protein interactions. Cellular localization at the light and electron microscopic level has also greatly contributed to this analysis.
Cell culture systems have also served as a valuable tool to understand the effects that disease-associated mutations have on protein localization, structure, and function. Typically, wild-type and mutant plasmids are used to transfect HEK293, COS7, or CHO cells for the transient or stable expression of proteins. Mutant protein expression and localization is determined by Western blotting and immunofluorescence microscopy for comparison with the wild-type protein. The extent of expression provides a reliable indicator of the stability of mutant proteins relative to wild-type proteins. Mutant proteins may express at exceedingly low levels (<10%) relative to the wild-type protein. This may reflect severe protein misfolding, mislocalization, and rapid degradation. Mutants that express at sufficient levels (>50%) are suitable for analysis. Immunofluorescence microscopy using cell-specific markers provides a means for comparing the localization of expressed mutant protein with wild type (24). If a high-quality antibody is not available, then it is useful to insert an epitope tag or green fluorescence protein fusion protein at one end of the protein. In most cases, membrane proteins are synthesized in the endoplasmic reticulum (ER) of the cell and transported to their cellular location via vesicle trafficking mechanisms. Mutations can affect the folding of the protein or the targeting sequence, resulting in mislocalization. Indeed many mutated membrane proteins are misfolded and retained in the ER by its quality control system and hence exhibit a markedly different distribution than the wild-type protein (13,21,24). ER localization can be confirmed using a specific ER marker such as calnexin. In some cases, a protein may be only partially misfolded and hence show a cellular distribution characteristic of both the wild-type and mutant protein (25). Some proteins containing ER retention sequences may be retained in the ER even as wild-type proteins, as in the case of ELOVL4, an enzyme associated with very long–chain fatty acid elongation (26). It should be noted, however, that the localization of proteins overexpressed in culture cells may not necessarily reflect their localization in situ. Hence, it is important to combine these heterologous expression studies with immunocytochemical labeling in retina tissue or other suitable natural cell sources to more definitively define their true cellular localization.
Culture cell expression is also useful for analyzing the functional properties of wild-type and mutant proteins. In some cases, the function of a membrane protein can be analyzed in extracts of cells expressing the protein. Alternatively, it may be necessary, if not preferable, to solubilize and purify the proteins for direct functional analysis or for reconstitution into vesicles (27,28). The purification of membrane proteins can be achieved using epitope tags such as a 1D4 or FLAG tag. Comparison of the functional properties of the mutant proteins with the wild-type proteins can provide important insight into the effect of mutations on the structure and function of the proteins
In the following sections, analysis of two pediatric retinal diseases, Stargardt macular degeneration and XLRS, is described with respect to their clinical phenotypes, genetic variants, localization, and protein analysis. Other aspects of these conditions can also be found in Chapter 19 (Stargardt Disease) and Chapter 27 (XLRS).
STARGARDT MACULAR DEGENERATION
Disease Characteristics
Stargardt disease (STGD1; MIM #248200) is the most common form of early-onset macular degeneration with an incidence of 1:10,000. It was first described in 1909 by the German ophthalmologist Karl Stargardt as a retinal disease displaying yellowish flecks in the central retina and atrophy of the macula (29). In 1960s, Franceschetti described a patient with similar flecks in the central retina and called the disease fundus flavimaculatus (30,31). Subsequent clinical and genetic studies have shown that Stargardt disease and fundus flavimaculatus are the same disease (32). Fundus flavimaculatus is now considered as a late-onset variant of Stargardt disease.
Stargardt disease is an autosomal recessive disease with symptoms most often appearing in the first or second decade of life (31,33,34). Affected individuals typically experience significant loss in central vision with a marked reduction in visual acuity. Progressive loss in visual acuity typically occurs throughout life with values reaching 20/200 to 20/400 or greater in later stages of the disease. A delay in dark adaptation and variable loss in color vision is often observed in Stargardt patients (34,35).
Ophthalmoscopic examination of Stargardt patients typically shows bilateral atrophy in the macula associated with the degeneration of photoreceptor cells and underlying RPE cells. Yellow-white flecks at the level of RPE cells are present in the macula often extending out to the midperiphery of the retina. A dark choroid is observed by fluorescein angiography in the majority of patients and results from the absorption of blue excitatory light by lipofuscin (31). Scotopic and photopic ERGs vary widely with some Stargardt patients showing relatively normal full-field ERGs and others displaying a significant loss in scotopic and/or photopic ERGs. A significant loss in photoreceptor cells and RPE cells in the macula is evident in donor eyes from deceased Stargardt patients with the remaining RPE cells displaying an abnormal appearance with excessive accumulation of lipofuscin (36).
ABCA4 Gene and Disease-Associated Mutations
The gene for autosomal recessive Stargardt disease was first mapped to chromosome 1p21–p13 and subsequently identified through positional cloning by Allikmets et al. (37) in 1997. It consists of 50 exons and encodes a 2,273 amino acid ATP-binding cassette (ABC) transporter. The gene for the Rim protein, a relatively abundant high molecular weight photoreceptor glycoprotein first described in the late 1970s (38), was cloned at the same time and found to code for the same ABC transporter associated with Stargardt disease (39). The gene formerly known as ABCR is now called ABCA4.
ABC transporters comprise a superfamily of proteins found in all prokaryotic and eukaryotic organisms (40). They utilize the energy of ATP binding and hydrolysis to transport a diverse group of substrates across cell membranes including nutrients, drugs, antibiotics, vitamins, lipids, amino acids, inorganic ions, polypeptides, and polysaccharides. ABC transporters typically consist of two transmembrane domains (TMDs) and two ABCs or nucleotide-binding domains (NBDs). Most eukaryotic transporters are either full transporters in which all four domains are located on a single polypeptide chain or half transporters in which one TMD and one NBD are present on a single polypeptide chain, which assemble as a functional homo- or heterodimer.
Human ABC transporters have been organized into seven subfamilies (ABCA–ABCG) based on similarities in gene organization and sequence. ABCA4 is the fourth identified member of the 12-member ABCA subfamily. ABCA4 exhibits a high degree of sequence identity (35% to 50%) and similar structural organization to other ABCA proteins (41). It is most similar to ABCA1, a widely expressed ABC transporter implicated in cholesterol and phospholipid efflux from peripheral tissues and linked to Tangier disease and familial high-density lipoprotein deficiency (42).
Over the past 15 years, molecular geneticists have screened Stargardt patients throughout the world for disease-causing mutations in ABCA4 (10,37,43–46). Over 800 mutations are now known to cause Stargardt disease. These include missense, nonsense, splice site, frameshift, and small deletion and insertion mutations. Of these, missense mutations are the most common and found throughout the coding region of the gene (Fig. 25.3). The amino acid substitutions p.G1961E, p.A1038V, and p.G863A appear with relatively high frequency in North America and Europe, possibly arising from a founder effect. Genetic studies suggest that 2% to 3% of the general population are carriers having a disease-causing mutation in one allele.
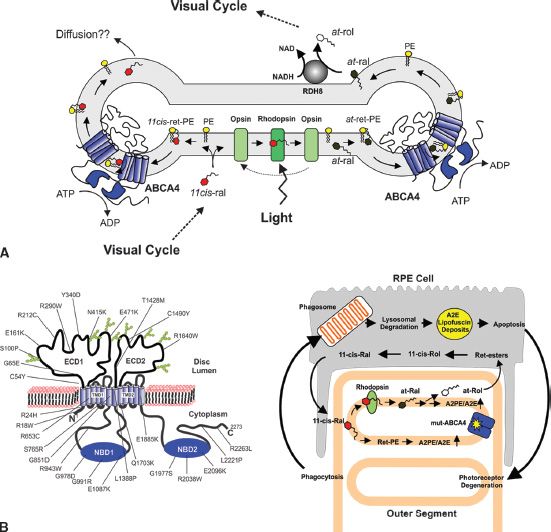
FIGURE 25.3 Proposed role of ABCA4 in retinoid processing and Stargardt macular degeneration. A: ABCA4 localized to the rim region of photoreceptor outer segments utilizes ATP binding and hydrolysis to flip N-retinylidene–PE from the lumen to the cytoplasmic side of the disc membrane, thereby facilitating the removal of retinal from disc membranes; B: (Left) Topological model for the organization of ABCA4 in the disc membranes. Mutations known to cause Stargardt disease are found throughout the protein; (right) proposed mechanism whereby mutations in ABCA4, which compromise its activity, result in the elevation of retinal and N-retinylidene–PE in disc membrane leading to the formation of A2E and its accumulation in RPE cells following phagocytosis of outer segments.
Mutations in ABCA4 have also been associated with other clinically severe retinal degenerative diseases. A homozygous 1 bp deletion in ABCA4 was first reported to be responsible for a form of autosomal recessive RP known as RP19 (47). This mutation, which causes a frameshift in codon 616 of exon 13 resulting in the addition of 32 amino acids prior to a premature stop codon, encodes a severely truncated protein, which is most likely unstable and rapidly degraded in the cell. A consanguineous family in which affected members had either a RP or CRD phenotype has also been reported (48). Family members with the RP phenotype exhibited chorioretinal atrophy, central scotomas, and a marked reduction in visual acuity as well as night blindness, restricted peripheral visual fields, attenuated retinal vessels, bone spicules, and reduced ERGs. Individuals with CRD showed severe loss in central vision together with some loss in peripheral vision. Genetic screening studies have now shown that mutations in ABCA4 account for up to 60% of autosomal recessive CRD.
Mutations in ABCA4 have also been implicated in some forms of AMD (49). Several studies have shown that individuals heterozygous for certain Stargardt disease–causing mutations in ABCA4 have an increased risk of acquiring AMD. In particular, two mutations p.G1961E and p.D2177N have been found to be present in individuals with AMD at statistically higher numbers than controls.
It is now generally believed that mutations in ABCA4 result in a spectrum of related retinal dystrophies, the severity of which depends on a number of factors. These include the type of mutations and its effect on protein expression and cell stress, localization, function, age of diagnosis, stage of the disease, and genotypic variations of individuals. In some cases, individuals first diagnosed with Stargardt disease can progress to the more severe phenotypic variant CRD (46).
ABCA4 Gene and Protein Expression
ABCA4 shows a restricted tissue distribution with high gene expression in the retina and little if any in the lung, liver, spleen, kidney, heart, and brain (37). In the retina, ABCA4 expression is limited to photoreceptors with the protein localized to the light-sensitive photoreceptor outer segment layer. Immunofluorescence labeling studies have further shown that ABCA4 is present in the outer segments of foveal and peripheral cone as well as rod photoreceptor cells (50). Immunoelectron microscopy studies have further localized ABCA4 to the rim and incisures of rod and cone photoreceptor outer segment discs (38,39,51).
Structural Features of ABCA4
ABCA4 is a relatively abundant 250kDa membrane glycoprotein that comprises up to 5% of the membrane protein by weight. It is highly conserved across vertebrate species, with the human protein being over 88% identical in sequence to other mammalian orthologues and 66% identical to the lower vertebrate such as Xenopus laevis (41).
Like other full ABC transporters, ABCA4 is organized in two tandem halves with each half containing a TMD followed by an NBD. Each TMD consists of six membrane spanning segments. A large exocytoplasmic domain (ECD) separates the first transmembrane segment from a cluster of five transmembrane segments in each half. The TMDs from both the N and C halves of ABCA4 form the substrate binding cavity and pathway for substrate translocation across the membrane. The ECDs contain multiple N-linked glycosylation sites and intramolecular disulfide bonds, which are important in the proper folding and stabilization of the native protein (52).
The NBD in the N and C half of ABCA4 each consisting of approximately 200 amino acids are 37% identical in sequence to each other. They contain the characteristic protein structural folds present in other ABC transporters including Walker A and Walker B motifs, which flank the signature C motif (53). In addition, ABCA4 contains a short 24 amino acid positively charged N-terminal segment that is important in determining the orientation of ABCA4 during protein biosynthesis and a 140 amino acid C-terminal segment that is crucial for the proper folding of ABCA4 into a native protein.
ABCA4 Functions as an N-Retinylidene–PE and PE Importer
Several lines of evidence have implicated ABCA4 in the transport of N-retinylidene–phosphatidylethanolamine (PE), a Schiff base adduct of retinal and phosphatidylethanolamine (PE), across disc membranes. ABCA4 purified from rod outer segment membranes and reconstituted into PE-containing lipid vesicles has a basal ATPase activity that is stimulated several fold by the addition of all-trans or 11 cis-retinal, but not retinol or retinylesters (27,54). Solid-phase binding studies indicate that N-retinylidene–PE binds with high affinity to purified ABCA4 and is released by the addition of ATP (55). More recently, an assay has been developed to measure retinoid transport of ABCA4 from disc membranes or reconstituted proteoliposomes to liposomes (28). ATP-dependent transfer of retinal was observed to occur with the flipping of N-retinylidene–PE from the lumen to the cytoplasmic side of disc membranes as the rate-limiting step. The basal ATPase activity of ABCA4 was also shown to be responsible for the flipping of PE in the same direction, a function that may contribute to the PE lipid asymmetry observed in disc membranes. Most well-characterized ABC transporters function as exporters translocating their substrates from the cytoplasmic to the extracellular or lumen side of a cell membrane. Interestingly, ABCA4 is unique in that it is the only mammalian ABC transporter and one of only several eukaryotic ABC transporters identified to date that functions as a substrate importer.
Characterization of Abca4 Knockout Mice
Abca4 knockout mice have provided valuable insight into the role of ABCA4 in photoreceptor cells. At a biochemical level, Abca4 knockout mice were first reported to have an elevated level of PE in retinal photoreceptor cells and show a light-dependent increase in all-trans retinal and N-retinylidene–PE (17,56). A light-dependent accumulation of the diretinylidene pyridinium compound A2E, also known as N-retinylidene–N-retinylethanolamine, was also observed along with a buildup of lipofuscin deposits in the RPE cells. The photoreceptor cells appeared normal with little if any sign of degeneration over the 1st year of life when reared under normal lighting conditions. Mild alterations in retinal morphology, however, were observed when Abca4 knockout mice were exposed to intense light for a short period of time (57). The ERGs and visual responses are also in the normal range except for the rate of dark adaptation. In an initial study Abca4 knockout mice were reported to show a delay in dark adaptation similar to that observed in Stargardt patients (17). A subsequent study, however, indicated that abca4 knockout mice actually adapt at a faster rate than wildtype mice (58).
A double knockout mouse deficient in both ABCA4 and the retinol dehydrogenase, RDH8, has been generated and characterized to further evaluate the roles of these proteins in retinoid processing in disc membranes (57). These double knockout mice showed significant light-induced degeneration compared to either of the single knockout mice. Degeneration of the photoreceptors and RPE cells correlated with a decrease in clearance of all-trans retinal from photoreceptors and a marked increase in A2E and lipofuscin in the RPE cells.
More recently, the light-dependent increase in lipofuscin and A2E in Abca4 knockout mice has been challenged (59). No significant difference of lipofuscin or A2E levels was found when Abca4 (−/−) mice were reared in the dark or under cyclic lighting conditions, although Abca4 knockout mice exhibited elevated levels of lipofuscin and A2E compared to wild-type mice as previously reported (17). These and other studies have led to the conclusion that A2E and related retinoid compounds, which comprise a significant fraction of lipofuscin deposits, arise primarily from 11-cis retinal and not all-trans retinal (59).
Disease-Associated Mutations in ABCA4
The effect of Stargardt disease mutations on the expression, subcellular localization, and functional properties of ABCA4 has been analyzed in transiently transfected culture cells (25,28,60). Amino acid substitutions that introduced charged amino acid residues in putative TMDs and small in-frame deletions resulted in low ABCA4 expression presumably due to protein instability and rapid degradation. In contrast, many disease-causing missense mutations in the NBD, ECD, and C-terminal segments of ABCA4 express at levels comparable to wildtype ABCA4. These mutant proteins showed a significant loss in ATPase activity and ATP-dependent retinoid and phospholipid transport as well as an altered cellular distribution indicative of protein misfolding. The targeting of ABCA4 mutants in transgenic Xenopus laevis photoreceptor cells has also been investigated by fluorescence microscopy. Several disease-associated ABCA4 mutants were retained in the inner segments of photoreceptors (61). Taken together, these studies indicate that the loss in functional activity as well as mislocalization of ABCA4 contribute to the pathogenic mechanisms underlying Stargardt disease and may be responsible in part for the wide range of phenotypes displayed by Stargardt patients.
Models Depicting the Role of ABCA4 in Photoreceptor Cells and Stargardt Disease
Biochemical studies showing that ABCA4 functions as N-retinylidene–PE transporter together with analyses of Abca4 knockout mice and Stargardt patients have led to proposed models depicting the possible role of ABCA4 in the physiology of photoreceptor cells and the pathogenesis of Stargardt disease. In the initial model, ABCA4 was implicated in the removal of all-trans retinal from photoreceptor disc membranes following photobleaching of rhodopsin or cone opsin (17,27,53). All-trans retinal released from rhodopsin after photoexcitation can either be directly reduced to all-trans retinol by RDH8 or react with PE in the disc membrane to form N-retinylidene–PE, a portion of which is trapped on the luminal side of the disc membrane. ABCA4 is envisioned to flip N-retinylidene–PE from the lumen to the cytoplasmic leaflet of the disc membrane. Upon dissociation of N-retinylidene–PE, all-trans retinal can be reduced by RDH8 and enter the visual cycle for the resynthesis of 11-cis retinal and regeneration of photoreceptor pigment. This mechanism is proposed to facilitate the complete removal of all-trans retinal from disc membranes following photoexcitation, thereby preventing condensation side reactions that form precursors of the diretinal compound A2E. Mutations that cause a loss in ABCA4 activity as an N-retinylidene–PE transporter would lead to an accumulation of all-trans retinal and N-retinylidene–PE in disc membranes and initiate the production of A2PE. Phospholipase D present in photoreceptor and RPE cells hydrolyzes A2PE to A2E and phosphatidic acid, with A2E accumulating with other diretinoid and lipid compounds as fluorescent lipofuscin deposits in RPE cells (62). A2E and related diretinal compounds can adversely affect RPE cell survival leading to RPE and photoreceptor cell degeneration. Support for this model has come from the initial findings that Abca4 knockout mice display a light-dependent increase in all-trans retinal and N-retinylidene–PE in photoreceptor cells and a light-dependent increase in A2E and lipofuscin in RPE cells (17,63) and biochemical studies showing that N-retinylidene–PE derived from all-trans retinal and PE activates the ATPase activity of ABCA4 and is transported from the lumen to the cytoplasmic side of disc membranes (27,28).
More recent studies have shown that lipofuscin and A2E accumulation in abca4 knockout mice is independent of light exposure and the rate of all-trans retinol production in abca4 knockout mice is the same as wildtype mice (59,64). These studies argued against a primary role of all-trans retinal in the formation of A2E in Abca4 knockout mice and Stargardt patients. Instead, a model has been proposed in which ABCA4 functions in the transport of the 11-cis form of N-retinylidene–PE from the cytoplasmic to the lumen leaflet of the disc membrane (59). This export direction, however, is counter to the import direction experimentally determined for ABCA4 (28). Furthermore, this model would result in an accumulation of 11-cis retinoid compounds in disc membranes in wild-type mice harboring ABCA4, leading to an increase in A2E formation.
A different model can be devised that incorporates the more recent findings that N-11-cis-retinylidene–PE is the preferred substrate for ABCA4 and the import direction of transport as shown in Figure 25.3. In this model 11-cis retinal, which enters photoreceptor outer segments from the visual cycle, preferentially interacts with opsin to regenerate rhodopsin. Excess 11-cis retinal, however, will react with PE to form N-11-cis retinylidene–PE or alternatively undergo isomerization to its all-trans isomer that can be trapped on the lumen side of the disc membrane. ABCA4, which can utilize either the all-trans or cis isomers of N-retinylidene–PE as substrate, is envisioned to transport N-retinylidene–PE to the cytoplasmic side of the disc membrane. Upon dissociation, 11-cis retinal not utilized for the regeneration of rhodopsin can be removed from disc by simple diffusion possibly facilitated by the localization of ABCA4 along the rim region of the disc membranes. Since RDH8 cannot reduce 11-cis retinal, other RDHs located in other parts of the photoreceptor cells and retinal cells may detoxify 11-cis retinal by reduction to 11-cis retinol. If isomerization occurs, then all-trans retinal can be reduced to all-trans-retinol for entry into the visual cycle. Hence, the disease phenotype of Stargardt patients may occur primarily as a light-independent increase in 11-cis retinal and its derivatives in photoreceptor outer segments due to a deficiency in ABCA4. Although ABCA4 may play a primary role in the removal of 11-cis retinal from photoreceptor cells, it may also play a role in the removal of all-trans retinal generated from the photobleaching of rhodopsin under extreme conditions including intense light exposure, limiting NADPH, or a deficiency in RDH8 activity. The more severe phenotype observed in abca4/rdh8 double knockout mice compared to rdh8 single knockout mice supports a role of ABCA4 in the clearance of all-trans retinal under such conditions. ABCA4 may also play an auxiliary role in generating PE asymmetry via its active transport of N-retinylidene–PE and PE (28).
Therapeutic Approaches
At the present time, there are no effective treatments for Stargardt disease. However, knowledge of the genetics and molecular and cell biology of Stargardt disease has led to the exploration of therapeutic interventions in animal models and initial clinical trials. In one approach, retinoid compounds have been investigated for their capacity to inhibit enzymes of the visual cycle, thereby reducing the level of 11-cis retinal in photoreceptors and correspondingly the production of A2E. Isotretinoin or 13-cis retinoic acid (also known commonly as Accutane for acne treatment) has been shown to inhibit 11-cis RDH, the enzyme which catalyzes the conversion of 11-cis retinol to 11-cis retinal in the visual cycle. A significant decrease in A2E levels and lipofuscin deposits was observed in Abca4 knockout mice given isotretinoin. This treatment was accompanied by a reduction in 11-cis retinal consistent with the effect of isotretinoin on the biosynthesis of 11-cis retinal via the visual cycle (65). Since isotretinoin exhibits adverse side effects, other retinoid compounds have been examined. These include retinylamine, an inhibitor of RPE65 isomerase, N-(4-hydroxyphenyl) retinamide also known as fenretinide, a retinoid compound that lowers serum levels of vitamin A, and various primary amines (57,66,67). These compounds generally show a reduction in A2E accumulation in animal studies.
Gene replacement therapy is another therapeutic approach under development. Since Stargardt disease is a recessive disorder, delivery of the normal gene to photoreceptors harboring the mutant gene should prove effective. Recombinant adeno-associated viral (rAAV) vectors are the vehicle of choice to deliver genes to photoreceptors and RPE cells since they are nontoxic and nonimmunogenic and can transduce nondividing cells for long-term protein expression. However, rAAV vectors have a limited DNA packaging size of 4.8 kb, considerably less than the 6.8 kb size for the coding region of ABCA4. Efforts are underway to develop dual vector protocols in which the gene is split into two overlapping pieces, each of which is packaged into separate AAV vectors. Co-injection of these vectors could lead to the translation of the full-length protein or two halves of the protein that could associate to form an active complex, although this remains to be demonstrated. Another approach involves using adenoviral or lentiviral vectors, which have a larger DNA packaging size. A study using lentiviral vectors harboring ABCA4 is currently in phase I clinical trial for Stargardt disease by Oxford BioMedica UK Ltd. To date, no data have been provided to determine the safety and efficacy of this treatment.
X-LINKED JUVENILE RETINOSCHISIS
Disease Characteristics
XLRS, first described by the Austrian ophthalmologist Josef Haas in 1898, is a leading cause of juvenile macular degeneration in males with a worldwide prevalence ranging from 1:5,000 to 1:25,000 (68). Affected children typically experience reading difficulties associated with a loss in visual acuity in the first decade of life. However, children as young as 3 months of age have been diagnosed with the disease suggesting that XLRS is a congenital disease (69). Stationary or slow disease progression is often observed in mid-years with visual acuity in the range of 20/50 to 20/100, although considerable variation is apparent even for affected individuals within the same family. Atrophy of the macula and retinal pigment abnormalities develop in the later stages of the disease resulting in a further reduction in vision (70–73).
As the name implies, a characteristic feature of XLRS is a splitting or schisis of the retina. Foveal schisis is observed in all patients with cystic streaks radiating from the foveal region of the retina in a spoke–wheel pattern. In about 50% of the cases, bilateral schisis is observed in the peripheral retina. Secondary complications can occur, including retinal detachment in about 10% of the cases and vitreous hemorrhages in about 4% of the cases, often resulting in a poor visual outcome (71).
The negative full-field ERG is another characteristic feature of XLRS (73,74). The dark-adapted “scotopic” response shows a marked reduction in the b-wave amplitude with only a minor change in the a-wave amplitude, implicating an abnormality of signal transmission involving bipolar cells. Significant reduction in the light-adapted “photopic” response is also evident, indicating that cone as well as rod photoreceptor signaling pathways are dysfunctional in the disease. Since the ERG response varies considerably in XLRS patients and the loss in b-wave amplitude is seen in other retinal diseases such as congenital stationary blindness, ERGs are not always an accurate diagnostic measure of the disease (72). OCT, which can noninvasively image the retinal layers, is now more widely used for the diagnosis and assessment of XLRS (74–77). Cystic cavities in XLRS patients are often present in several layers of the retina including the inner retinal and nerve fiber layers as visualized by OCT. Small cystic cavities in the parafoveal region often coalesce near the fovea into a large fluid-filled cavity. Older patients often show a marked reduction or absence of schisis cavities and a thinning of the retina. Heterozygous female carriers are clinically asymptomatic unlike other X-linked retinal degenerative diseases such as X-linked RP and X-linked choroideremia. However, minor alterations in the ERGs have been reported (77).
RS1 Gene and Disease-Associated Mutations
The gene responsible for XLRS (RS1; OMIM #312700) was first identified in 1997 by Sauer et al. (78) using positional cloning and analysis of retina-specific transcript expression. The gene maps to position Xp22.13 on the short arm of the X-chromosome and spans 32.4 kb of genomic DNA. Six exons encode a 224 amino acid protein known as retinoschisin or RS1. Retinoschisin contains an N-terminal signal or leader peptide, which targets proteins to the ER lumen, and a discoidin (DS) domain. Both these structural features are found in a number of extracellular proteins and cell surface membrane proteins (79).
Over the past 15 years, more than 191 unique disease-causing mutations in the RS1 gene have been reported in various populations throughout the world (Leiden Open Variation Database, LOVD version 2.0, Build 31; http://grenada.lumc.nl/LOVD2/eye/home.php?select_db=RS1). These include missense mutations (~52%), splice site mutations (29%), deletions (13%), duplications (3%), insertions (1.5%), and insertion/deletions (1.5%) (72). Forty percent of the mutations, including frameshift and nonsense mutations, are predicted to yield null alleles. Missense mutations are found throughout the protein, with a large majority being present in exons 4 to 6 encoding the DS domain (Fig. 25.4). The p.E72K (p.Glu72Lys) mutation is particularly common, being reported in over 66 families. Other relatively common mutations include p.R102Q, p.R102W, p.R141C, p.P192S, p.R200C, and p.R213W.
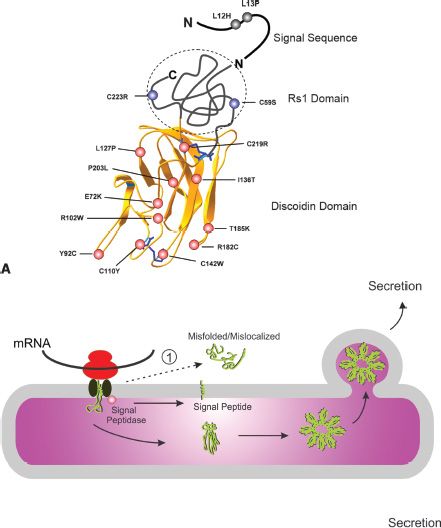
FIGURE 25.4 Mechanisms associated with X-linked retinoschisis. A: Model for retinoschisin showing the proposed structure of the DS domain and the location of the Rs1 domain and signal peptide sequence for which the structure has not been determined. Selected disease-associated mutations are shown in the DS domain (red), Rs1 domain (blue), and signal sequence (black). Disulfide bonds are shown in blue. B: Mechanism by which disease-associated missense mutations in RS1 cause nonfunctional proteins. Wild-type protein is synthesized off of ribosomes and threaded through the ER membrane. The signal peptide is cleaved off by a signal peptidase. The subunit folds into a native structure and assembles into an octameric complex via disulfide bonds. The native octamer is secreted from cells. Mutations in the signal peptide prevent the polypeptide from threading through the ER membrane; as a result the protein is misfolded and mislocalized to the cytoplasm where it is rapidly degraded. Mutations in the Rs1 domain allow proper folding of the polypeptide chain, but prevent subunit assembly into octamers. The protein gets secreted, but due to defective octamerization it is nonfunctional, resulting in the disease. Mutations in the DS domain cause protein misfolding and retention in the cell by the ER quality control system.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


