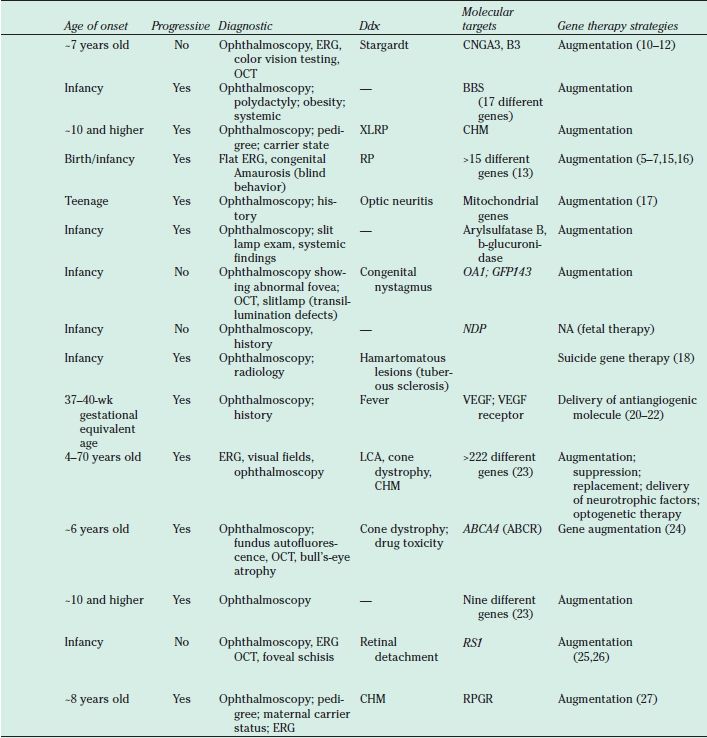
Listed are the diseases, the genetic defects, and the gene therapy strategies under consideration. ABCA4, ATP-binding cassette transporter cassette A member 4 gene; AR, autosomal recessive; NA, not applicable; AD, autosomal dominant; BBS, Bardet-Biedl syndrome; CHM, choroideremia; CNGA3, cyclic nucleotide gated channel alpha 3; CNGB3, cyclic nucleotide gated channel beta 3; Ddx, differential diagnosis; ERG, electroretinogram; GPR143, G protein-coupled receptor 143; LCA, Leber congenital amaurosis; LHON, Leber’s hereditary optic neuropathy; MPS, mucopolysaccharidosis; OA1, ocular albinism type 1; PR, photoreceptor; ROP, retinopathy of prematurity; RP, retinitis pigmentosa; RPGR, retinitis pigmentosa GTPase regulator; RS1, retinoschisin 1; VEGF, vascular endothelial growth factor; XL, X-linked; XLRP, X-linked retinitis pigmentosa. At present, there is no approved treatment for the retinal degenerative conditions. The role of physicians/health care providers is supportive, to provide a clinical and molecular diagnosis and to inform patients about the status of research developments.
Treatment of pediatric retinal diseases presents special issues not encountered with adult disease. The biology of many pediatric retinal conditions either involves or impacts normal development of the retina and the visual system. Changes that occur prenatally, such as the lack of formation of the fovea in ocular albinism or presence of a coloboma, are fully manifest at birth. Treatment performed after birth would be expected to have limited impact on this condition because the biologic effects are irreversible. In utero treatment would provide an opportunity to correct the defects before the signs of disease become manifest and thereby allow the development of an anatomically normal and functional retina. There are other blinding conditions that are detectable early in life but in which the cellular anatomy is roughly intact. However, in diseases that develop pathologic changes postnatally, such as retinopathy of prematurity (ROP) or familiar exudative vitreoretinopathy, there can be a window of time for intervention after birth to prevent permanent loss of foveal fixation and abnormal development of central visual pathways with amblyopia.
Although the technical demands of in utero intervention would present major challenges, there would be distinct advantages. Prenatal application of an exogenous gene would help induce immune tolerance to the foreign antigen. Transduction efficiency would likely be enhanced because a larger area of the outer retina could be targeted with one injection. This is not only because of the greater number of cells present per unit area in the fetal retina but also because there are no attachments between the neural retina and the retinal pigment epithelium (RPE) early in fetal life since the two layers do not interdigitate until photoreceptor outer segments are formed. The natural space between these cell layers prior to differentiation permits free diffusion of the subretinally administered drug. Correction of the defect at an early stage would also reduce the occurrence of later tissue damage. This is important in early-onset retinal degeneration because photoreceptors, which die in these diseases, are terminally differentiated cells after birth and cannot regenerate. Finally, early application of the therapeutic gene would take advantage of the plasticity of the developing retinal to central nervous system (CNS) axonal connections present early in life. If treatment were performed too late in development, higher order CNS neurons might already have established synapses from other sensory systems, and cortical areas for vision would no longer exist.
Several retinal diseases exhibit a short, well-defined period of biologic activity in the early postnatal period and early childhood. Intervention would need to occur during or before infancy in order to arrest the disease process and salvage retinal function. For example, threshold ROP occurs on average about 37 weeks’ postgestational age. Retinal detachment as stage 4 ROP typically occurs about the time of what would have been a full-term delivery, 40 weeks’ gestation (Table 24.1). The window of opportunity for treatment in ROP is thus limited to a period of a few weeks. In inherited retinoblastoma, tumor appearance usually occurs within the 1st year of life, sometimes presenting at birth. Effective treatment for retinoblastoma would require intervention within a narrow time period of active tumorigenesis. In LCA, visual deficits are present at birth or in early childhood. The retina undergoes progressive degeneration through childhood and adult life. Intervention with gene therapy should ideally take place within a defined time period before histologic degeneration of photoreceptors and retina develops. Results from a gene augmentation therapy clinical trial for LCA due to RPE65 mutations indeed show that, although subjects of all ages show improved vision after gene therapy, children treated with gene therapy attain higher levels of retinal/visual function than adults (5,28).
Several retinal diseases exhibit a short, well-defined period of biologic activity in the early postnatal period and early childhood. Intervention would need to occur during or before infancy in order to arrest the disease process and salvage retinal function. For example, threshold ROP occurs on average about 37 weeks’ postgestational age. Retinal detachment as stage 4 ROP typically occurs about the time of what would have been a full-term delivery, 40 weeks’ gestation (Table 24.1). The window of opportunity for treatment in ROP is thus limited to a period of a few weeks. In inherited retinoblastoma, tumor appearance usually occurs within the 1st year of life, sometimes presenting at birth. Effective treatment for retinoblastoma would require intervention within a narrow time period of active tumorigenesis. In LCA, visual deficits are present at birth or in early childhood. The retina undergoes progressive degeneration through childhood and adult life. Intervention with gene therapy should ideally take place within a defined time period before histologic degeneration of photoreceptors and retina develops. Results from a gene augmentation therapy clinical trial for LCA due to RPE65 mutations indeed show that, although subjects of all ages show improved vision after gene therapy, children treated with gene therapy attain higher levels of retinal/visual function than adults (5,28).
GENERAL PRINCIPLES OF GENE THERAPY
Gene transfer can be achieved by several different methods (Table 24.2). Ultimately, the optimal technique will achieve the gene expression profile required for the specific application with the minimum of toxicity. Naked deoxyribonucleic acid (DNA) does not penetrate cell membranes readily; however, transient expression with modest transduction efficiency can be achieved by a variety of physicochemical techniques. The advantage of these techniques is there are no size limitations to the DNA, there is no exposure to potentially immunogenic virus capsid proteins, and it is possible to produce these vectors in large quantities. In some instances, electroporation can be used to deliver DNA to target cells (29,30). Transfer to deep tissue layers (of the cornea) can be achieved by ballistic injection of DNA bound to gold particles (gene gun) (31). Cationic lipids that interact with cell membranes can enhance transfer efficiency (32,33). With many of these physicochemical techniques, efficiency of transfer to many cell types and duration of gene expression are quite limited. One promising approach that appears to prolong the duration of expression involves the use of agents to condense the DNA (Table 24.2) (34,35).
TABLE 24.2
Vectors under evaluation for human ocular gene therapy
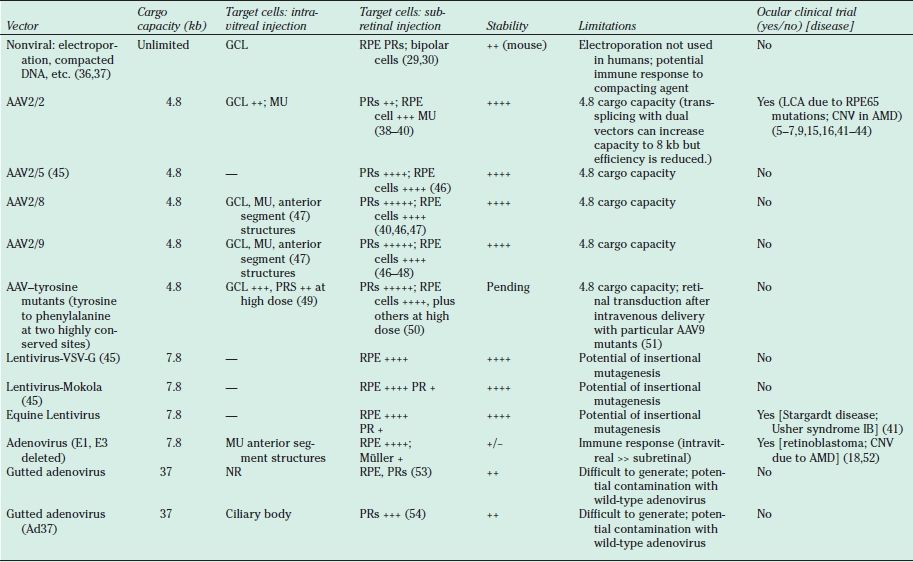
Transduction characteristics and potential for use in pediatric retinal diseases. Only viruses that are capable of transducing postnatal retinal cells are listed. Viruses that have been or are being used in approved human clinical trials are indicated under column entitled “Clinical Trial.” References are not all-inclusive except for clinical trial reports but are intended as examples. AAV, adenoassociated virus; VSV-G, vesicular stomatitis virus G protein; AMD, age-related macular degeneration; kb, kilobases; NR, not reported; RPE, retinal pigment epithelium; PR, photoreceptors; GCL, ganglion cell layer; MU, Müller cells; LCA, Leber congenital amaurosis; CNV, choroidal neovascularization.
Viruses are used to take advantage of their ability to transfer exogenous nucleic acids into host cells. Viral vectors have been considered ideally suited for treatment of many diseases because of their superior gene transfer efficiency and stability of gene expression. Using recombinant DNA engineering techniques, viruses are designed so that their ability to replicate is eliminated and their pathogenic properties are minimized. Different viruses demonstrate distinctly different characteristics with regard to cargo capacity, cell tropism, immune response, and stability of transgene expression (Table 24.2). Of the 1,786 human gene therapy clinical trials that have been carried out through 2011, the vast majority (74.2%) have used recombinant viruses as gene delivery vectors (55). Of these human gene therapy trials using recombinant viruses, adenovirus has been used in the most protocols (55).
Recombinant adenovirus is highly efficient at transducing RPE cells and Müller cells (56,57). Onset of gene expression is rapid, occurring within 24 to 48 hours of vector administration. However, transduction efficiency with adenovirus of retinal cell types such as photoreceptors is poor. In addition, the duration of gene expression is limited by the lack of integration of the transgene and the host immune response to the vector (58,59). Adenoviruses were used in phase 1 clinical trials for retinoblastoma and for choroidal neovascularization secondary to age-related macular degeneration (Table 24.1).
A significant improvement in gene transfer technology involves generation of “gutted” or “helper-dependent” adenoviruses, viruses in which all viral open reading frames have been removed. These viruses have a large (~38 kb) cargo capacity and also have reduced immunogenicity, as evidenced by the fact that transgene expression persists longer than is observed with earlier generation adenoviruses (60). It is possible to modify the cellular targeting characteristics of such viruses by changing envelope components (54). Widespread evaluation of gutted adenoviruses remains to be done because these viruses are very difficult to grow and often are contaminated with wild-type virus.
Inflammation from systemic delivery of a viral vector can result in severe toxicity. The most dramatic example, which occurred during a human gene therapy trial, resulted in the death of a human subject (61). Injection of the early-generation (E1- and E3-deleted) recombinant adenoviral vectors into the vitreous also can induce profound inflammatory responses. After high-dose recombinant adenovirus delivery to the eyes of rodents, inflammatory panophthalmitis can be seen. The degree of inflammation is markedly less when the vector is administered into the subretinal space. This difference in immune response has been attributed to the suppression of the inflammatory response when the vector is placed in the immunologically privileged environment of the subretinal space (59). In addition, injection into the subretinal space reduces the exposure of viral vector to uveal tract structures that are in contact with the vitreous space (e.g., ciliary body, iris). In the human ocular gene therapy trial involving retinoblastoma, the recombinant adenovirus was used to deliver the cDNA encoding thymidine kinase of the herpes simplex virus to children with retinoblastoma. Treatment with ganciclovir is then used to kill the tumor cells (so-called suicide gene therapy) (62). Immune response directed at the adenovirus vector might contribute to the therapeutic effect in this study.
Retroviruses have long been available for use in gene therapy applications. Many retroviral vectors appear to have limited application in retinal gene therapy due to their inability to efficiently target nonreplicating, terminally differentiated cells, such as those in the neural retina. Retroviral vectors have prolonged expression, in part due to the fact that this vector integrates into the host genome (63). Although this might be an advantage, it also poses a risk of insertional mutagenesis. This risk was realized in a human gene therapy trial for X-linked severe combined immune deficiency, in which children whose disease had been “cured” by retroviral treatment developed a leukemia-like syndrome (64).
There is one class of retrovirus, lentivirus, which shows favorable somatic transduction profiles. The majority of the recombinant lentiviruses that have been evaluated are derived from the human immunodeficiency virus. Multiple safeguards are in place to minimize the risk of producing a wild-type (replication competent) virus during the development of recombinant lentiviral vectors. Recombinant lentiviral vectors have also been generated using nonhuman parental strains, such as those derived from the cow, cat, and horse. A phase I/IIa study using an equine-derived lentiviral vector for Stargardt disease has recently been initiated for adult subjects (Table 24.1) (55). A phase I/IIa study using a similar vector for treatment of the RP in Usher syndrome (type1B, MYO7A gene) will start imminently. Onset of expression of human-derived lentivirus occurs within 24 to 48 hours of administration and is seen primarily in the RPE (65,66). Chimeric lentiviral vectors have been engineered by packaging the genetic material from the lentivirus in the envelope of another virus (e.g., the rabies virus Mokola envelope). Altering the envelope can modify the tropism of the vector, altering the range of cell types that the vector will efficiently transduce.
In recent years, there has been a large increase in interest in the development of adeno-associated virus (AAV) as a vector for ophthalmic gene therapy. AAV has been used in 86 different protocols to date, approximately 6 of which involve the eye. Half of these are currently enrolling children with LCA due to RPE65 mutations (41).
AAV is a parvovirus and has minimal pathogenetic effects in humans and other animals. Although wild-type AAV can integrate into genomic DNA, integration has not been reported after delivery of recombinant AAV to the retina. Instead, the AAV-delivered transgenes appear to remain stable as episomes in the host nucleus, thereby providing stable gene expression over time. AAV has expression profiles that appear particularly favorable for applications to retinal disease. Dozens of different AAV serotypes have been identified or engineered, and many of these transduce photoreceptor cells with extremely high efficiency (38–40,50,66,67). Most of the different AAV serotypes have a strong tropism for the RPE. When injected into the vitreous, some AAV vectors are capable of transducing retinal ganglion cells (68). Although AAV does induce an immune response, the response is relative mild even after injection into the vitreous (5,69). The most significant limitation to the use of AAV is the small cargo capacity of the vector. Whereas recombinant adenovirus vectors can accommodate transgenes up to 7.8 kb in size, AAV can package a maximum of 4.8 kb (Table 24.2) (70,71). Use of dual AAV vectors can overcome this limitation to some extent (see later). Another potential limitation of AAV in therapeutic applications is the prolonged latency period prior to onset of gene expression that is seen with some AAV serotypes (39,66). Depending on the animal species treated and the serotype of the capsid, the delay to peak levels of transgene expression after vector administration can be up to 8 weeks (72). A corresponding delay in the expression of the transgene can miss the therapeutic window because the active pharmacologic product is absent during the peak period of disease activity. This situation has proven to be important in treating several different animal models of early-onset retinal degeneration.
Oxygen-induced Retinopathy Mouse
Numerous animal models exist for various pediatric retinal conditions, thus providing the opportunity to test gene therapy approaches for these diseases (Table 24.2). One well-established model for oxygen-induced retinopathy is the hyperoxia–hypoxia rodent model (73) (see also Chapter 43). In this model, neonatal mice are exposed to a very high oxygen tension for several days and then returned to normal oxygen levels. Neovascularization develops transiently at the junction of vascularized and avascularretina. The degree of neovascularization can be quantified, thus providing measures of retinopathy in these animals.
Several experiments have used this model to test antiangiogenic strategies in retinal neovascularization. AAV (and other) vectors containing different antiangiogenic compounds, including soluble vascular endothelial growth factor receptor-1 (sFlt-1), were injected in the early neonatal period, prior to exposing animals to high oxygen (20,74–78). Transgene expression occurred relatively soon after intraocular injection, thus exposing areas of potential neovascularization to antiangiogenic transgene products (4,5,7,75–77). The neovascularization that occurred in these eyes with retinopathy was reduced, often dramatically, without apparent deleterious effects to the native retinal vasculature (78). Using this model, gene therapy–mediated delivery of antiangiogenic compounds thus appears to be a viable approach for primary or adjuvant treatment of conditions with intravitreal neovascularization. A clinical trial using AAV2-mediated delivery of sFlt-1 has been initiated based on the fact that this approach appeared safe, localized, well tolerated, stable, and efficacious in large animal models (79,80). If shown to be safe and effective, the same strategy could potentially be applied to juvenile-onset diseases with intravitreal neovascularization (Table 24.2).
Spontaneous and Genetically Engineered Animal Models of Retinal Degeneration
RPE65 Mutant Animals
Several animal models exist for inherited retinal degenerations caused by specific gene mutations. Some of these diseases manifest at or soon after birth, whereas others result in early-onset retinopathies that are rapidly progressive. Abnormalities in the gene encoding the RPE-specific RPE65 protein can result in lack of photoreceptor function that is evident at birth. In humans, RPE65 mutations are responsible for as much as one-fifth of all cases of LCA (81,82). LCA can also be caused by mutations in other genes. Likewise, RPE65 mutations can lead to other human phenotypes. A naturally occurring homolog of lack-of-function RPE65 mutations occurs in Swedish Briard dogs in which an LCA-like syndrome of congenital nystagmus, blindness, and significantly depressed photoreceptor function occurs (83,84). An Rpe65 spontaneous knockout mouse exists (85) and a genetically engineered RPE65 knockout has been developed in mice (86). These animals (Rpe65−/− mice) suffer from a similar LCA-like syndrome.
Gene therapy using AAV carrying a functional RPE65 transgene has been used successfully to treat the canine and murine models of LCA caused by lack of RPE65 (85,87–92). In the canine disease, when the AAV vector is injected into the subretinal space (Figs. 24.1 and 24.2), animals with abnormal electroretinogram (ERG) responses from birth developed a normal-appearing ERG with amplitudes corresponding to the area of the retina treated and displayed visual behavior not previously present, and nystagmus eye movements were significantly reduced (87–90,93). Intravitreous injection of vector results in no expression of RPE65 in the outer retina and no corresponding development of ERG or visual response.
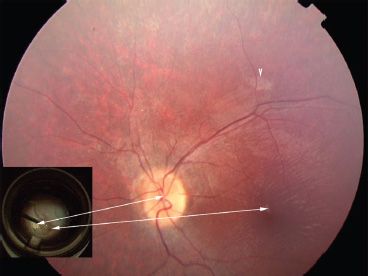
FIGURE 24.1 Fundus photo of a 14-year-old with LCA due to RPE65 mutations 2 weeks after subretinal injection of AAV2.hRPE65v2 in the follow-on study of the phase I/II gene therapy clinical trial at the Children’s Hospital of Philadelphia. The only sign that the retina has been injected is the retinotomy scar (arrowhead) superior to the macula. There is an ILM reflex typical for an LCA patient this age. The inset shows a still photo from the video taken during surgery. Arrows indicate the location of the optic disc and the fovea in the surgical photo compared to the postinjection fundus photo. A drop of high-density Perfluoron is layered on the fovea (and extends to the optic disc), protecting this structure during the injection. Just above the optic disc, one can appreciate the border of the localized retinal detachment caused by subretinal injection of the AAV.
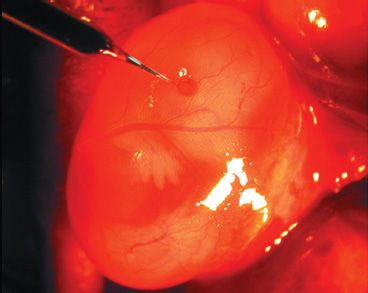
FIGURE 24.2 AAV was injected subretinally in one eye of a fetal mouse on embryonic day 14. (From Surace E, Auricchio A, Reich S, et al. Delivery of adeno-associated viral vectors to the fetal retina: impact of viral capsid proteins on retinal neuronal progenitor transduction. J Virol 2003;77:7957–7963, with permission.)
The compelling safety and efficacy data from all three animal models were used in 2007 to obtain institutional and national regulatory approvals to carry out three near-simultaneous clinical trials (Table 24.1)—a trial designed for adults in the United Kingdom (Bainbridge, PI, Moorfields Eye Hospital, London), a trial designed for children in the United States (Maguire, PI, Children’s Hospital of Philadelphia, CHOP), and a trial designed for adults in the United States (Jacobson, PI, University of Pennsylvania and University of Florida, Gainesville). These trials (plus one more that reported results from one patient [15]; a fourth trial at the University of Oregon Health Sciences Center has not published data as of yet) have provided complementary data sets revealing the high level of safety of subretinal delivery of AAV2 carrying the wildtype RPE65 cDNA in this disease (5,6,9,42,43,94). In addition, both subjective and objective measures of retinal/visual function have demonstrated efficacy stable over years in the two studies that have so far been reported in full (5,16). Data reported by Maguire et al. indicate that although all subjects benefited from the intervention, the extent of recovery of retinal and visual function in children is greater than in adults. Recovery of function was documented by a number of different noninvasive subjective and objective assays. In addition, functional magnetic resonance imaging (fMRI) studies documented that correction of the retina allowed the brain “to see” (28). The fMRI test results, like the results from the tests that had been previously used, showed that there was a greater recovery of vision in children than in adults. This result was expected given the data from the preclinical animal studies (87,93,95) and also from the fact that there is a degenerative component of this disease (resulting in fewer treatable retinal cells as subjects get older). The results of the fMRI studies also indicated that the neuronal connections between the retina and the brain were intact in the adults who were treated, even though they had been severely visually impaired for decades (7,28). This issue is important with respect to the ability to resuscitate vision later in life in other early-onset blinding disorders.
The phase I/II studies for LCA due to RPE65 mutations will continue, as mandated, through the 15-year time point for the studies that are in progress. In the meantime, it was not known whether the fellow eye could be safely treated in subjects who have already benefited from a single eye injection. The concern was that an immune response generated due to exposure of the virus capsid/RPE65 transgene product would lead to inflammation and prevent benefit after a second exposure. To evaluate the safety of second eye readministration, the CHOP team carried out large animal studies in unaffected nonhuman primates and in affected canines. These studies showed a high degree of safety and also of efficacy (90). Therefore, the CHOP team initiated a “follow-on” study evaluating the effects of readministration. The initial results from the first three subjects show a high degree of safety and efficacy (7). The reversal of blindness was documented by a number of tests in this study, including fMRI.
The LCA-RPE65 studies mark the first successful readministration of a gene augmentation therapy for an inherited disease. The success of this approach is likely due in large part to the favorable immunologic properties of the eye. These results have helped to formulate the plan for the CHOP team to proceed to phase III testing of gene therapy for LCA due to RPE65 mutations (anticipated start date in the first quarter of 2013) (41). This will be the first phase III study of gene therapy for an ophthalmic disease. The rapid progress for this disease is due in large part to the availability of the precious animal models.
Other Early-Onset/Rapidly Progressive Retinal Degeneration Animal Models
There are several models of early-onset RP. The spontaneous mutant mouse model of autosomal recessive (AR) RP due to lack-of-function mutations in the gene encoding cGMP rod photoreceptor–specific phosphodiesterase (PDEβ has been studied for at least half a century, and the Irish Setter dog model (rcd1) of this disease has been studied for more than two decades. There have been several reports of successful slowing of the disease process in the murine models of PDEβ-based disease. Success has been achieved through the delivery of wild-type PDEβ cDNA in a variety of viral vectors (Table 24.2) (53,96–98). Unfortunately, rescue in all of these reports was transient, apparently due to the overwhelming apoptotic cell death that takes place in this disease. It might be that studies in the rcd1 dog, which has a longer window of opportunity for treatment, will lead to a longer period of rescue. Alternatively, in utero treatment of the murine models could be successful.
There is a murine model of Stargardt disease, which is a juvenile-onset macular degeneration caused by mutations in the ATP-binding cassette transporter gene (ABCR, also known as ABCA4). This Abca4−/− mouse has many of the features of Stargardt disease, except that macular structure cannot be assessed because the mouse lacks a macula (99). Treatment of Stargardt disease by gene replacement therapy presents several problems. The ABCA4 cDNA is quite large, on the order of 8 kb. This poses limitations as to the type of vector that can be used to carry this gene in one piece (Table 24.1). Only lentivirus or gutted adenovirus can accommodate this gene; a gene therapy clinical trial for Stargardt disease run by Oxford BioMedica recently commenced using a lentiviral vector after carrying out preclinical testing (Tables 24.1 and 24.2) (24,41). An alternative strategy for this gene would involve splitting the cDNA in two at a natural splicing site, and each piece would be carried in by two different AAV vectors. This approach has been demonstrated using the reporter gene lacZ (100).
Both murine and canine animal models are also available for studies of another severe and early-onset form of RP: X-linked RP due to mutations in the gene encoding retinitis pigmentosa GTPase regulator (RPGR) (101,102). Recently, partial success in treating a canine model of this disease was reported in three of four affected dogs using AAV5-mediated subretinal delivery of the full-length RPGR cDNA (27). This brings hope that additional studies will be carried out that lead to gene therapy clinical trials for boys with this severe, early-onset disease.
Mouse models exist for another juvenile-onset X-linked retinal degeneration, choroideremia, and there has been good progress in developing a therapy for this disease (103). There is one report of lentivirus-mediated delivery of function in the choroideremia model (104). Although there are no publications as yet of AAV-mediated rescue of the retinal phenotype in this model, a clinical trial using AAV2-mediated gene augmentation therapy has been initiated for choroideremia (105). Because this disease progresses relatively slowly, it may take many months or years to determine whether there is any efficacy.
There are a number of other genetic forms of pediatric-onset retinal degeneration that are being evaluated for proof of concept of gene augmentation therapy studies in the United States and in Europe. These include forms of LCA caused by LCA5, AIPL1, CEP290, RDH12, and GUCY2D mutations. Autosomal dominant RP due to rhodopsin mutations is a target for gene suppression/augmentation therapy (106), and several groups are evaluating strategies for limiting or preventing disease caused by mitochondrial mutations in Leber hereditary optic neuropathy (LHON) (Table 24.1) (17,107,108).
Lysosomal Storage Diseases
Lysosomal storage diseases have also been identified as a potential gene therapy target. A variety of storage diseases exist in which there is an abnormal accumulation of substrate in a variety of tissues leading to multisystem disease, including retinal disease. The mucopolysaccharidoses (MPSs) are a group of such diseases characterized by the abnormal accumulation of glycosaminoglycans in the lysosomes as a result of defects in carbohydrate metabolism (109). Ocular abnormalities include corneal clouding, retinal degeneration, optic atrophy, and glaucoma.
Numerous animal models of MPSs exist and include (i) a feline model of MPS VI (deficiency of arylsulfatase B) and (ii) murine, feline, and canine models of MPS VII (deficiency of β-glucuronidase) (110,111). These models share multiple pathologic abnormalities with humans, including those seen in the eye.
Gene therapy has been successful in animal models by treating the ocular manifestations of MPS VII and MPS VI (112). Injection into the vitreous or into the corneal stroma of the adenovirus carrying the human β-glucuronidase gene resulted in complete clearance of the storage defect in MPS VII RPE and corneal cells (113,114). Intravenous administration of a recombinant AAV encoding the human β-glucuronidase cDNA also resulted in nearly complete elimination of lysosomal storage vacuoles in the RPE of these animals (115). For MPS VI, Ho et al. (116) used an AAV to deliver arylsulfatase B to the subretinal space of the MPS VI cat. AAV treatment reversed the disease phenotype in the RPE.
Retinoblastoma
Gene therapy studies have focused on developing an effective eye-sparing treatment for retinoblastoma. Current protocols use enucleation to treat large unilateral tumors in patients. This is often used in conjunction with chemotherapy or radiotherapy. Such procedures can be cosmetically destructive for pediatric patients. The goal of gene therapy is to eliminate the need for such drastic treatments and preserve the integrity of the eye. The availability of animal models for retinoblastoma has facilitated such gene therapy studies (Table 24.2).
Two murine models of human retinoblastoma have been developed: one metastatic and one nonmetastatic. In the first model, cells from the human-derived Y79 retinoblastoma cell line were injected into the vitreous of nude mice. These cells form intraocular tumors in the vitreous cavity, and the tumors progressively invade the retina, subretinal space, choroid, optic nerve head, and anterior chamber of the eye. The tumors progress into the subarachnoid space and focally invade the brain, where they often migrate to the contralateral optic nerve. In the second model, WERI-Rb tumors were injected into the vitreous of nude mice. Tumor cells were localized in the eye with only anterior choroidal invasion at late stages (117).
Hurwitz et al. (62) used an adenoviral vector to administer the cDNA for the herpes simplex virus thymidine kinase gene (HStk) to experimental tumors in mice, and animals were subsequently treated with ganciclovir. Transduced cells were rendered susceptible to ganciclovir cytotoxicity, and tumor growth was inhibited. These results allowed for the approval of a phase I clinical trial for the treatment of retinoblastoma. This phase I study involving eight children showed that adenovirus-mediated delivery of HStk followed by ganciclovir can be safe and can contribute to the treatment of children with retinoblastoma tumor seeds in the vitreous (18).
Retinal Dysgenesis
Ocular Albinism: Retinal Dysgenesis and Abnormal Visual Tract Development
All forms of albinism lead to abnormal development of the retina and the visual tracts leading to the brain. Children with albinism have abnormal foveal development and poor fine visual discrimination. Although foveal abnormalities cannot be studied in the Oa1 mouse model (because mice lack a fovea/macula) (Table 24.2) (118), the Oa1-/- mouse manifests two features that are present in the human disease and might be amenable to correction through gene transfer: (i) the presence in the RPE of macromelanosomes, which appear shortly after birth and increase in number until postnatal day 7, and (ii) the misrouting of optic fibers at the chiasm. Gargiulo et al. (119) showed that AAV-mediated delivery of a normal copy of the disease-causing gene to the RPE could restore melanogenesis and ocular function postnatally in this mouse model. It may be necessary to deliver the cDNA even earlier in development in the human (i.e., in utero) in order to allow normal development of the fovea.
Other Gene Defects Affecting Retinal Development
Retinal dysgenesis can result from CRX, NR2E3, or CRB1 lack-of-function mutations (Table 24.2). Animal models for some of these diseases exist and are amenable to gene therapy studies. The crx knockout mouse demonstrates photoreceptor abnormalities early in life and severely impaired ERGs (rod more than cone) (120). The NR2E3 gene encodes a nuclear receptor. Lack of this gene product causes enhanced S-cone syndrome (see Chapter 10), a disorder of retinal cell fate (121). The phenotype in the rd7 mouse has been shown to result from an Nr2E3 mutation (122) and could potentially be used for gene therapy studies. Although mutations in the neural retina leucine zipper gene (NRL) can result in autosomal dominant RP, lack of function of Nrl in a mouse model has been shown to have a dramatic effect on retinal differentiation. Nrl-/- mice lack rod photoreceptors and have only “cod” (i.e., cone) mutations (123). Studies in this model could also lead to treatment of enhanced S-cone syndrome (124). Due to the early onset of this disease, it may be necessary to test intervention at fetal stages in mice (Figure 24-2) (125).
X-linked juvenile retinoschisis (XLRS) is another disease that evolves from abnormal retinal differentiation. Lack of the retinoschisis gene product results in separation of the retina at the level of the nerve fiber and ganglion cell layers. This results in cystic degeneration of the central retina. ERGs of affected males have preserved rod and cone photoreceptor systems but substantially reduced b waves, indicating loss of bipolar cell activity (126). Several groups have demonstrated proof of concept for gene augmentation therapy of XLRS (Table 24.1) (25,127,128). Similar to XLRS, Norrie disease can involve retinal schisis. The involved gene (NDP) has been identified and a mouse model generated, which should be useful in developing a gene-based therapy (129).
Mutations in the Crumbs homolog 1 (CRB1) gene have been shown to result in a variety of early-onset retinal disorders, including defective retinal lamination, LCA, and Coats-like exudative vasculopathy (130–132). A spontaneous mouse model, the rd8 mouse, may be useful in developing a gene-based treatment (133). Finally, complete lack of color vision, achromatopsia, can result from mutations in the genes encoding the subunits of the cone photoreceptor cGMP-gated cation channels, CNGA3 and CNGB3 (134). Cone vision has been restored in both a mouse model and a canine model of achromatopsia using subretinal delivery and AAV5-mediated gene augmentation therapy (10,11).
APPLICATION OF GENE THERAPY TO PEDIATRIC RETINAL DISEASES
Numerous conditions exist that may be amenable to intervention using a gene therapy approach (Table 24.2). Timing of intervention requires careful consideration if successful therapy is to be achieved. Many pediatric retinal conditions manifest at an early stage of development and therefore would require early postnatal treatment or even in utero therapy (Fig. 24.3). Although in utero therapy presents substantial technical challenges (Fig. 24.4), certain advantages may be afforded by the timing of intervention. Exposure of antigen at this early stage can induce tolerance to the foreign transgene, thus minimizing the immune response. Efficiency of gene transfer and a reduction in latency for gene expression in actively replicating retinal cells might be increased. In addition, wound healing might be improved because scar formation is minimized with fetal surgery.
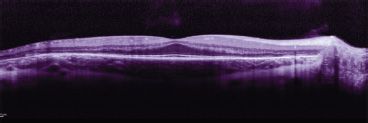
FIGURE 24.3 OCT: A potential future outcome measure for pediatric retinal gene therapy. OCT from this 14-year-old with choroideremia shows spared macular outer nuclear (photoreceptor) layer but significant thinning of this layer in the peripheral retina.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


