TABLE 23.2
Usher genes, proteins, and mouse models
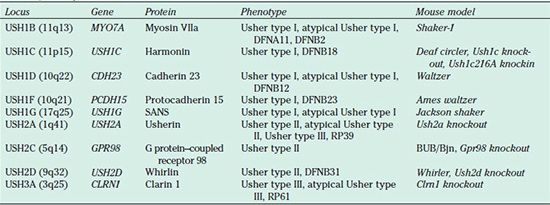
DFNA* and DFNB* designate forms of autosomal dominant and recessive nonsyndromic hearing loss, respectively, that are due to mutations in an Usher gene. RP designates nonsyndromic retinitis pigmentosa.
USH1A (14q32) and USH2B (3p23–24) are not listed in the table because follow-up studies demonstrated that they do not exist.
USH1E (21q21), USH1H (15q22–23), and USH1K (10p11–q21) have not yet been identified.
It is important to realize that a child with Usher syndrome is likely to be given the misdiagnosis of nonsyndromic sensorineural hearing loss with the correct diagnosis of Usher syndrome not entertained until the onset of visual loss. Thus, use of effective ophthalmologic and genetic diagnostic tools is critical for every child diagnosed with nonsyndromic sensorineural hearing loss because of the possibility that the child may have Usher syndrome. An early diagnosis provides the family and child with time to prepare for the physical, emotional, and educational impact of the approaching visual loss, following satisfactory management of the hearing loss.
More than 50% of the deaf–blind population and about 18% of individuals with retinitis pigmentosa have Usher syndrome (4). It is found in most ethnic groups, and prevalences ranging from 3.2 to 6.2 per 100,000 have been reported in numerous European countries as well as the United Kingdom, the United States, and Colombia. However, a recent genetic study of deaf and hard of hearing children living in the state of Oregon found that 11% had Usher syndrome, and it was estimated that the population prevalence may be as high as 1 in 6,000 (5).
Up to 75% of Usher patients have Usher syndrome type II, while the most severe form, Usher syndrome type I, accounts for 25% to 40%. The most frequent genetic subtypes are USH2A and USH1B. In general, Usher syndrome type III is rare with a frequency of ˂6% in most populations, but in the Finnish and Ashkenazi Jewish populations, 40% of Usher patients have type III (3).
As the major cause of deafness and blindness, Usher syndrome is an important public health problem, particularly if it is not diagnosed until the onset of visual loss. The importance of early diagnosis for effective management and intervention cannot be overemphasized, especially with the major ongoing advances in the development of therapeutic approaches to delay and/or stop the progression of the retinal degeneration.
In Usher syndrome type I, the hearing loss is severe to profound across all frequencies, and the onset of retinitis pigmentosa is during childhood (Table 23.1). Vestibular function is generally absent although some type I patients with normal response to bithermal vestibular testing have been reported (6). Hearing aids are usually not helpful, but cochlear implantation is proving to be beneficial for many individuals with Usher type I (7).
The degree of hearing loss in patients diagnosed with Usher syndrome type II increases from moderate in the low frequencies to severe in the high frequencies and tends to remain stable; carefully fitted hearing aids usually work well for them. They have normal vestibular function, and the age of onset of retinitis pigmentosa is usually during the teenage years. Usher syndrome type III is distinguished from the other two types by the progressive nature of the hearing loss. The age of onset of retinitis pigmentosa and the degree of vestibular dysfunction are variable (Table 23.1).
The development of pigmentary retinopathy differentiates Usher syndrome from nonsyndromic hearing loss, with night blindness often being the first indication of retinal degeneration. Vision loss gradually increases as a result of progressive degeneration of rod photoreceptor cells, which is followed by loss of the retinal pigment epithelium (RPE) layer, and may progress to total blindness by the third or fourth decade. Phenotype and rate of progression are quite variable among Usher patients; in fact, 25% may have normal structure and function in some regions of the retina (8). However, this variation does not provide criteria for differentiating the three clinical types, and electroretinogram (ERG) and optical coherence tomography findings are consistent with a shared pathologic mechanism (9).
The inheritance pattern in Usher syndrome is autosomal recessive, meaning that affected children have two abnormal copies of the same gene, one inherited from each parent. Males and females are equally likely to be affected, and the parents are carriers but show no signs of the disease. If both parents are carriers of mutations in the same Usher gene, then each child has a 25% chance of having Usher syndrome, and the probability that an unaffected child is a carrier is two-thirds. Note that a positive family history is not expected with an autosomal recessive disorder such as Usher syndrome because the abnormal copy of the gene may be passed on from one (unaffected) carrier to the next for many generations before a couple, who by chance both carry a mutation in the same Usher gene, has an affected child (Fig. 23.1; see also Chapter 15).
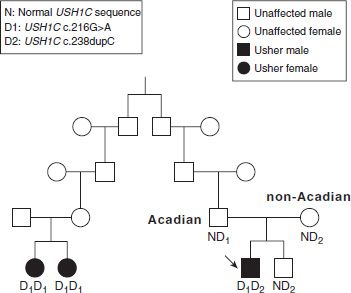
FIGURE 23.1 Pedigree of Usher syndrome type I family living in Acadiana. The parents of the affected male (designated by arrow) are carriers of two different USH1C mutations, D1 [c.216G>A] and D2 [c.238dupC], and the child is a compound heterozygote with the genotype D1D2. His unaffected sibling is a carrier of D2, and the affected female relatives both have the genotype D1D1.
Based on estimated population prevalences of the Usher syndrome genetic subtypes, the frequencies of carriers of mutations in each of the Usher genes can be estimated. For example, for a population in which 70% of Usher patients have Usher syndrome type II and 80% of those have USH2A, the frequency of carriers of mutations in USH2A is approximately 1 in 50 (assuming the prevalence of Usher syndrome is 1 in 6,000). Similar calculations for each of the Usher subtypes suggest that the total frequency of carriers of a mutation associated with Usher syndrome may approach 5% in some populations.
Although the clinical classification of Usher syndrome according to type can be helpful, genetic diagnostic testing has demonstrated that the correspondence between clinical findings and Usher type is by no means clear-cut. For example, mutations in MYO7A, CDH23, and USH1G have been found in individuals with clinical findings closer to type II or type III than type I, while genetic testing has detected USH2A mutations in some individuals with a clinical diagnosis of type III. Adding to the diagnostic dilemma is that some individuals with mutations in Usher genes (e.g., MYO7A, USH1C, CDH23, PCDH15, USH2D) have hearing loss but do not develop retinitis pigmentosa (they have nonsyndromic hearing loss, not Usher syndrome), while conversely some individuals with mutations in Usher genes (e.g., USH2A and CLRN1) develop retinitis pigmentosa but do not have hearing loss (they have nonsyndromic retinitis pigmentosa, not Usher syndrome). A database of reported mutations in Usher genes may be accessed using the following URL: https:// grenada.lumc.nl/LOVD2/Usher_montpellier/ USHbases.html
Approximately 55% of Usher type I patients have USH1B. It is caused by mutations in the MYO7A gene, which encodes the unconventional myosin VIIa protein (10), a member of the large superfamily of myosin motor proteins that move along cytoplasmic actin filaments in an ATP-dependent manner. Mutations in the orthologous mouse gene cause the deafness mutant Shaker-1, and the gene was identified through studies of this mutant (11).
MYO7A has 48 coding exons extending over approximately 87 kb of genomic sequence and it is expressed in several alternative splice forms. More than 220 different mutations have been reported in MYO7A with most of them having population frequencies of <0.04. They occur throughout the gene, but do tend to cluster in the exons encoding important conserved domains of the protein (12–14). While the majority of MYO7A mutations are found in USH1B patients, some are associated with autosomal recessive (DFNB2) and autosomal dominant (DFNA11) nonsyndromic deafness (Table 23.2).
Myosin VIIa is a 254-kDa protein consisting of 2,215 amino acids. As is typical of myosins, it has a motor head domain containing ATP- and actin-binding motifs and a long tail with two large repeats (FERM) of about 460 amino acids. It is expressed in the cochlear hair cells of the inner ear and both the RPE cells and the photoreceptor cells of the retina; it has also been detected in testis, lung, kidney, intestine, and olfactory epithelium. In all of these epithelial tissues, highest expression is associated with the cilia and microvilli of the apical surfaces. The photoreceptors require myosin VIIa for transport of opsin, while in the RPE cells it facilitates movement of melanosomes and phagosomes. In the inner ear, myosin VIIa may be essential for adhesion between the stereocilia of the cochlear hair cells.
Scanning electron micrographs of the cochlea of a Shaker-1 mouse show disorganization of the sensory hair bundles, which are normally highly organized structures with precisely arranged rows of stereocilia on the apical surface of the hair cells (15). Shaker-1 mice have mild ERG abnormalities but no apparent photoreceptor degeneration, although it has been reported in specific light/dark exposure conditions (16). Surprisingly, the retinas of Shaker-1 mice appear to be protected from acute light damage, suggesting that myosin VIIa has a role in the visual retinoid cycle. Localization and motility of melanosomes is defective in the RPE cells of these mice and opsin accumulates in the connecting cilium of the photoreceptors, which is consistent with a failure of transport to the outer segment (17).
USH1C was first described in the Acadian population of Louisiana; it accounts for up to 15% of Usher type I patients in most populations, but almost all who are of Acadian ancestry. The associated gene, USH1C, encodes a PDZ domain protein named harmonin, which probably functions as a scaffolding protein. PDZ domains are involved in organizing the assembly of protein complexes. The USH1C gene contains 28 coding exons spanning 51 kb of genomic sequence, and at least 10 harmonin isoforms, divided into three subclasses (a, b, c), are found as a result of alternative splicing of eight exons (18–20).
More than 20 USH1C mutations have been reported, most affecting splicing or located in an exon encoding a PDZ domain (12–14). The Acadian mutation introduces a cryptic splice site in exon 3 (c.216G>A). One copy of this mutation was detected in a profoundly hearingimpaired child born in 2000 to an Acadian father and a non-Acadian mother. Sequencing of the USH1C gene identified a second mutation (c.238dupC), thus establishing the correct diagnosis of USH1C. If the parents had not been aware of two distant cousins with a possible diagnosis of Usher syndrome (Fig. 23.1), it is unlikely that they would have pursued genetic testing, and the child would probably have been misdiagnosed as nonsyndromic bilateral profound hearing loss. Following the Usher syndrome diagnosis, the parents were proactive in obtaining a cochlear implant for their 12-monthold son. Mutations in USH1C have also been associated with nonsyndromic hearing loss (DFNB18).
The longest isoform of harmonin, b, is expressed in the hair cells of the developing inner ear, but has not been detected in the retina. It contains about 900 amino acids and three PDZ domains. Isoform a is the most common and also has three PDZ domains while isoform c has two; a and c have 400 to 500 fewer amino acids than isoform b. However, whereas isoform b expression is restricted to the inner ear, both a and c are present in most tissues, including all compartments of the neural retina (21).
A spontaneous mutant, Deaf circler, and targeted (Ush1c knockout and knockin) mouse models exist for USH1C (22–24). All of these mice are deaf and show similar hair cell disorganization to that seen in the Shaker-1 mouse. In addition, the knockin mouse model containing the Acadian c.216G>A mutation exhibits retinal degeneration (23).
USH1D is associated with mutations in a novel cadherinlike gene (CDH23) and is probably the second most common form of Usher type I (25,26). Mutations in this gene were also found in the mouse mutant Waltzer, in which the hair bundles are disorganized just as in Shaker-1 mice (27). CDH23 has 69 coding exons and spans more than 300 kb of genomic sequence; approximately 150 mutations have been reported (12–14). As with MYO7A and USH1C, mutations in CDH23 are also found in patients with nonsyndromic hearing loss (DFNB12). There is a correlation between phenotype and type of mutation, with missense mutations giving a less severe Usher phenotype than null mutations (e.g., nonsense, frameshift, splice site), and all observed DFNB12 mutations are missense (28).
The protein encoded by the CDH23 gene has 3,354 amino acids and 27 extracellular cadherin repeats. Cadherins are a large protein family, which contain extracellular calcium–binding domains and are involved in intercellular adhesion. Cadherin 23 makes up the upper part of the tip links of the hair cell stereocilia and directly binds to the tail of myosin VIIa. These two proteins together with harmonin form a ternary complex; thus, it is likely that myosin VIIa applies tension forces on the hair bundle links (29).
Mutations in another cadherin-like gene, PCDH15, were found in patients with USH1F, which accounts for about 11% of Usher type I (30). The murine homolog of this protocadherin gene was shown to be defective in the Ames waltzer mouse, in which the disorganized hair bundles are very similar to those in other Usher type I mouse models (31).
PCDH15 has 33 exons and spans close to a 1,000 kb of genomic DNA. At least 50 mutations have been reported (12–14), and one of these, p.Arg245X, accounts for the majority of USH1F cases in the Ashkenazi Jewish population (32). Additionally, large genomic rearrangements are a relatively frequent cause of USH1F (33). As with other Usher type I genes, mutations in PCDH15
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


