BBS, Bardet-Biedl syndrome; RP, retinitis pigmentosa; MKS, Meckel-Gruber syndrome; LCA, Leber congenital amaurosis.
Clinical Symptoms and Signs
The age of onset of BBS is at conception, when a fetus inherits two abnormal copies of one of the BBS genes. Typically one mutant allele is inherited from each parent, although uniparental disomy for an abnormal BBS gene or inheritance of one abnormal gene and spontaneous mutation of the other allele are also theoretical possibilities.
Polydactyly
The diagnosis of BBS may be suspected at birth if polydactyly is present. This may be manifest as extra fingers, toes, or both. The supernumerary digits are typically extraaxial, near the little finger or toe; however, there is a report of an extra finger between the third and fourth digits (19). Digits may be rudimentary or fully formed. A syndrome of isolated autosomal dominant polydactyly also exists and is relatively common, so extra digits may be ligated and removed shortly after birth leaving only tiny scars. Parents may not mention this on history, and patients may be unaware that extra digits were present at birth. For this reason, specific questions about extra digits should be asked, and hands and feet examined if BBS is suspected later in life.
Retinal Degeneration
In the absence of polydactyly, the first symptom of BBS is usually decreased vision in dim light, typically noticed around the age of 5 to 7 years. The electroretinogram (ERG) may be abnormal before this; in a large cohort of families with BBS, all patients with genetically proven BBS who were older than 3 years had some evidence of retinal degeneration (20). What begins as night blindness progresses to visual field loss and RP. Acuity is often good in the early stages of retinopathy, and since young children have limited independent activities in dim light at this age, specific questions must be asked to elicit symptoms. Questions such as “Can he/she find a seat in a dark movie theater without assistance?” and “Can he/she see stars at night?” are often illustrative. Parents may report noticing that the child has difficulty ambulating when camping at night or trick-or-treating on Halloween. In bright light the child may appear to see perfectly and normally. Nystagmus has been reported in 59% of patients (20), yet is not considered a typical finding in BBS. Since central vision loss is not present during the 1st months of life, the nystagmus is not sensory in origin. 43.7% of patients in one study developed cataracts (20). Most patients in this study were legally blind by late teens or early adulthood, but no patient lost all light perception. At a young age the fundus appears normal. Many patients develop a “bull’s-eye maculopathy” appearance, which may begin as a blunted fovea (Fig. 22.1). The peripheral vision decreases steadily, but central vision is retained longer. When central vision does decrease, it may be attributable to cystoid macular edema (CME) and/or epiretinal membranes. Over time, atrophy of the macula occurs (Fig. 22.2). On slitlamp examination of the anterior vitreous in childhood, a significant cellular reaction can be seen (Fig. 22.3). This may precede a decline in vision (Drack, unpublished data, 2012). The cells seen in the vitreous may be an inflammatory or autoimmune overlay related to the retinal degeneration. Posterior subcapsular cataracts may develop over time, probably related to the chronic cellular reaction. Legal blindness develops by the time patients are in their 20s or 30s due to severely decreased field, maculopathy causing poor acuity, or both. A typical RP picture with waxy pale disc, thinned arterioles, and bone-spicule–like pigmentation often develops at this point (Fig. 22.4). In mouse models of BBS a striking loss of outer retinal layers is seen on histology (Fig. 22.5).
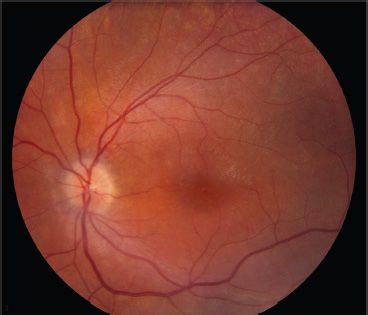
FIGURE 22.1 Left fundus of a 10-year-old with BBS1. Visual acuity was 20/70 right and 20/40 left. The child had been followed since age 3 years for nystagmus, esotropia, and amblyopia as well as obesity. Referral was made when nyctalopia was noted.
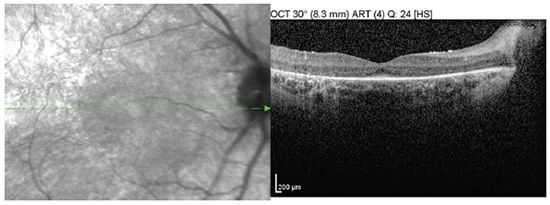
FIGURE 22.2 Red free and OCT of the right fundus shows subtle bull’s-eye appearance and thinning of outer retinal layers.

FIGURE 22.3 Anterior vitreous cellular reaction in the right eye of the same patient.
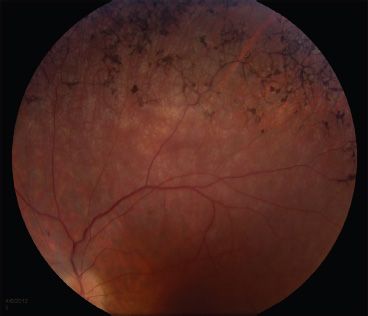
FIGURE 22.4 Bone-spicule–like pigmentation in the retinal periphery of a 15-year-old with BBS
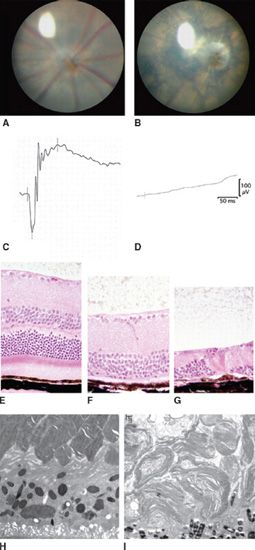
FIGURE 22.5 Summary of BBS mouse model ocular phenotypes characterized by our research group. Images from mutant mice (B,D,F,G,I) are compared to normal wild-type (WT) controls (A,C,E,H). (A) shows a WT mouse retinal fundus. (B) shows fundus abnormalities including arteriolar constriction, scalloped hypo- and hyperpigmentation, and pale optic nerve in a Bbs3−/− mouse; (C) shows a normal ERG response in a WT mouse. (D) demonstrates ERG loss in a 5-month-old BBS mouse; (E–G) show normal histology of the retina in a WT mouse in the first panel and progressive retinal degeneration in BBS mice retinas at two points in time (F,G); ultrastructural abnormalities in BBS mouse OSs (I) compared to control retina (H).
Obesity
Obesity may precede vision loss but is rarely suspected to be due to BBS unless an astute clinician searches for other features or recalls polydactyly. The obesity is truncal. While infants with BBS are usually of normal weight, many children are noticeably obese by 1 to 5 years of age. The obesity, which accompanies BBS, is especially difficult to manage. Studies in mice and humans have demonstrated that it is due to leptin resistance (5,21). Leptin is a hormone that triggers a feeling of satiety, so resistance renders those afflicted with BBS not feeling full after eating a sufficient quantity of food. In addition, animal studies have shown that even when eating the same amount of food as unaffected littermates, BBS mice gain more weight (5), suggesting that energy expenditure also plays a role.
Developmental Disability
Developmental delay and psychiatric disorders are overrepresented in BBS patients but are not universal. In one study, patients with BBS due to mutations in BBS1 or in BBS12 appeared to have milder cognitive disorders with several having university degrees (20). In a behavioral study of 21 BBS patients, repetitive behaviors, obsessions, and autistic features were noted, but none had diagnosable autism. Anxiety, depression, and difficulty with social interactions and with attention were also noted (22). BBS mice exhibit a social dominance phenotype (23).
Other Features
Later occurring complications include diabetes mellitus, renal insufficiency, gonadal insufficiency, and hypertension. One study suggests that BBS10 patients may have the most severe renal disease (20). Some patients have heart defects. Anosmia is a frequent finding, although patients may not be aware of it since it is present from early life; BBS2 mice also have anosmia (23,24). Hearing loss and “glue ear” (serous otitis media) have been reported in some BBS patients. Surveillance for these disorders should commence in childhood and the importance of close follow-up should be discussed.
Diagnostic Studies
When BBS is suspected based on ocular history or physical findings, a prudent first step is an ERG. This is a challenging test for many children to perform, especially if they have developmental or behavioral issues as is the case in many with BBS. Using Dawson Trick Litzkow microfiber electrodes rather than contact lens electrodes may make an otherwise unobtainable examination possible. If an ERG is not possible while the child is awake, ERG with sedation or anesthesia should be considered, keeping in mind that ERG amplitudes may be variably reduced by up to 50% by anesthetic agents. Sedation and anesthesia have a small but significant risk of complications. Therefore, if the clinical picture is typical and ERG cannot be performed easily, doing genetic testing before ERG is advisable. The ERG in BBS early in the course is usually a rod–cone dystrophy pattern. Later the ERG often becomes nonrecordable (see also Chapter 10 on Electrophysiology).
Genetic Testing
At this writing 70% to 80% of patients will have a molecular diagnosis on genetic testing (2). Certainty of diagnosis is important to rule out other causes of retinal degeneration in children and to put into place the proper surveillance for systemic disorders. In addition, since this is an autosomal recessive disorder, genetic counseling for recurrence risk is important for families. Clinical laboratories testing for BBS can be found at www.genetests.org. At this writing it is available for most of the 17 genetic types. Many patients have novel mutations, so complete sequencing of all of the known BBS genes is often necessary. A certified genetic counselor or geneticist should be consulted to help patients and families understand the implications of genetic testing results and to counsel about recurrence risk.
All patients who can cooperate should have a slitlamp biomicroscopic examination to look for cells in the anterior vitreous and subsequent posterior subcapsular cataract. If central acuity is diminished, optical coherence tomography (OCT) is often helpful to diagnose CME.
Communication with geneticists and primary care doctors is imperative since these patients will need lifelong follow-up for systemic complications. Referral to a low-vision specialist will aid in advising the school about the child’s visual needs and will help adults adapt to their reduced vision. Some patients may have vision adequate for driving in the teenage years, but since the retinal degeneration is progressive, this should be monitored closely. Referral to nutritionists and educational specialists is also recommended. An endocrinologic evaluation around puberty is also beneficial.
BBS has variable expressivity. Some patients have been reported with retinal degeneration alone and no other features of BBS. These patients were detected in families with BBS3 and BBS9 in which some members have typical multisystem BBS and other family members have isolated RP and share the same two BBS mutations (25). Some patients have a milder, more slowly progressive retinal degeneration than the norm. For this reason, it is important to counsel families and prognosticate based on the clinical examination of the individual patient, rather than to tell parents and patients that all of the many possible complications are inevitable.
Pathophysiology
At the cellular level, BBS is associated with ciliary dysfunction and pathogenic defects are found in ciliated cells. Insights into the pathophysiology of BBS have been gained through the use of animal models in which specific BBS genes have been disrupted. For example, mutation of BBS genes in mice results in absence of flagella in spermatozoa and abnormalities in cilia in brain ependymal cells, airway epithelial cells, and olfactory neurons (16,17,23,24). The photoreceptor cell outer segment (OS), which captures light and initiates the downstream signal transduction cascade in the retina, is a highly specialized form of the primary cilium, and the photoreceptor cells degenerate in human BBS patients and BBS animal models (4). At the molecular level, BBS proteins are involved in protein trafficking between the ciliary and plasma membranes. Among the known BBS proteins, seven (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, and BBS9) form a stable complex, known as the BBSome (26) (Fig. 22.6). The BBSome associates with the intraflagellar transport particles, which are essential for cilia and flagella formation, and mediates protein trafficking along the microtubules (27). BBS3 (also known as ARL6) is a member of the Ras superfamily of small GTPases and controls BBSome recruitment to the membrane and BBSome ciliary entry (28). BBS6, BBS10, and BBS12 show sequence homology to the CCT/TRiC family of group II chaperonins (29–32). These three BBS proteins form another complex (the BBS/CCT chaperonin complex) together with six CCT chaperonins and mediate BBSome assembly (33). BBS13 (MKS1) and BBS14 (CEP290) localize to the transition zone at the ciliary base and perform a “gatekeeper” function, controlling the entry and exit of ciliary proteins (34–36). Mutations in the MKS1 and CEP290 genes are also associated with other cilia-related genetic diseases, Meckel-Gruber syndrome (MKS), Joubert syndrome (JBTS), and Senior-Loken syndrome (SLSN), which share several phenotypic features with BBS (35,37–40). BBS15 is caused by mutations in WDPCP; mutations in this gene can also cause MKS. This gene is important in cellular movement during embryogenesis and ciliogenesis (41). BBS16 is caused by mutations in SDCCAG8/NPHP10 and reportedly does not cause polydactyly (42). BBS17 is caused by mutations in LZTFL1 and is associated with a specific type of insertional polydactyly (19).
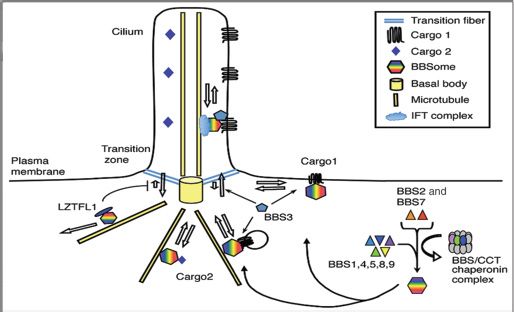
FIGURE 22.6 Current working model for BBS protein functions. Seven BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8, and BBS9) form the BBSome that functions as a coat complex to sort cargos and mediate ciliary trafficking. BBS6, BBS10, and BBS12 form the BBS/CCT chaperonin complex together with CCT family chaperonins and mediate BBSome assembly. BBS3 recruits the BBSome to the membrane and facilitates its ciliary entry. LZTFL1 binds to the BBSome in the cytoplasm and inhibits BBSome ciliary entry. The BBSome is thought to associate and cooperate with the IFT complex within the cilia. (Modified from Sheffield VC. The blind leading the obese: the molecular pathophysiology of a human obesity syndrome. Trans Am Clin Climatol Assoc 2010;121:172–181.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


