FIGURE 21.1 Pigmentary retinopathy vessel attenuation.
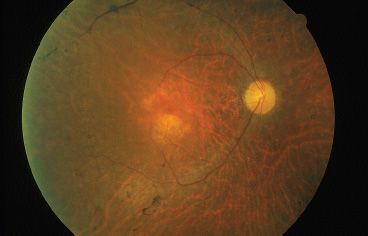
FIGURE 21.2 “Waxy pallor” of optic nerve in autosomal dominant retinitis pigmentosa. Note narrowed arterioles.
RP may occur in isolation or as part of a multiorgan syndrome. This provides a convenient method of division; we will divide the discussion of the pigmentosa into nonsyndromic RP and syndromic RP. Although syndromic RP is noted in the presence of systemic neurologic or metabolic diseases, in nonsyndromic disease, the eyes are affected in isolation. Syndromic RP should be excluded by a thorough history, physical examination, and appropriate laboratory studies as some patients require nonocular clinical intervention. Certain inflammatory and infectious conditions affecting the retina and choroid can mimic the RP fundus findings. Drug toxicity can produce an RP-like fundus picture, along with old ophthalmic artery occlusion. These pseudo-RP conditions should be excluded by history and appropriate evaluation. To explore each of these entities, this review first examines the nonsyndromic forms of RP and correlates these diseases with recent genetic information. This review explores pseudo-RP conditions, their clinical and laboratory features, as well as syndromic RP conditions presenting with neurologic and metabolic diseases.
NONSYNDROMIC RETINITIS PIGMENTOSA
RP most often occurs as an isolated ocular condition. RP conditions are caused by genetic defects in the visual phototransduction cascade, RPE and vitamin A metabolism, transcription factors, and structural defects in photoreceptors. Because of the genetic heterogeneity of these defects, the multiple mutations within each individual gene, and the possibility of digenic inheritance, there is great phenotypic variability. This creates variability in age of presentation, inheritance, and phenotype. In this chapter, we discuss typical RP, retinitis punctata albescens, and conditions that mimic RP.
Clinical Symptoms and Signs
Isolated RP represents a majority of patients with inherited retinal disease. Patients present with difficulty or clumsiness in dim light. Questions eliciting information about activity in low light or nighttime can reveal nyctalopia. The classic findings of RP include nyctalopia, bone spicules in the midperiphery, attenuated arterioles, waxy pallor of the optic disc, VF constriction, and a depressed scotopic and later photopic ERG patterns. Early clinical examination might only reveal subtle changes including RPE mottling, a fine layer of intraretinal pigment, clumping of pigment in the retinal layers, focal RPE atrophy, and mild attenuation of vessels (Fig. 21.2). Even at this early stage, there can be substantial ERG abnormalities. A diminished scotopic response, diminished combined photoreceptor response, and a delayed implicit time are typical.
Isolated RP can be subdivided into three common inheritance patterns: XL, AR, and AD. Cases without a known inheritance pattern are referred to as simplex. Most simplex cases of RP reflect AR inheritance or incomplete penetrance. Along with these inheritance patterns, digenic transmission (via two AR genes) has also been documented. As of 2011, six XL RP genes have been mapped, and three of these have been cloned. Forty recessive loci have been mapped, and 37 of these have been cloned. There have been 22 AD RP loci mapped, and 21 of these have been cloned. There have also been two forms of digenic and one form of mitochondrial transmission reported, but these were in syndromic RP.
Prevalence/Incidence
The overall incidence of RP in the United States has been calculated to be approximately 1:3,700, thus making RP the most prevalent of all retinal dystrophies (3). The reported proportions of each pattern of inheritance vary, with a US study estimating 10% AD, 84% AR (including simplex cases), and 6% XL (3), whereas a UK study found approximately 20% AD, 70% AR, and 10% XL (4). The highest recorded prevalence of RP is 1:1,800 in the Navajo of the southwestern United States (5). Simplex cases can account for 50% of all cases. AR inheritance is presumed to account for most of these cases. Incompletely penetrant AD RP, new AD RP mutations, and digenic and XL RP are purported to account for a small percentage of these cases.
Autosomal Dominant Retinitis Pigmentosa
AD RP typically presents in the second to the fourth decades and is often the mildest form of the disease. It may be unrecognized because of its slow progression, subtle clinical findings, and late onset. Reduced penetrance may give the illusion of “generation skipping.” Variable penetrance may also occur, particularly in RP9 (PAP1), PRPF31, and RP1. Dominant RP is thought to account for 10% to 25% of all RP cases. Proteins responsible for AD RP include rhodopsin, peripherin/ retinal degeneration slow (RDS), neural retina leucine zipper (NRL), cone–rod homeobox (CRX), retinal fascin (FSCN2), precursor RNA–processing complex C (PRPC8), and precursor RNA–processing factor 31 (PRPF31) (Table 21.1).
TABLE 21.1
Proteins, genes, and loci identified in nonsyndromic retinitis pigmentosa
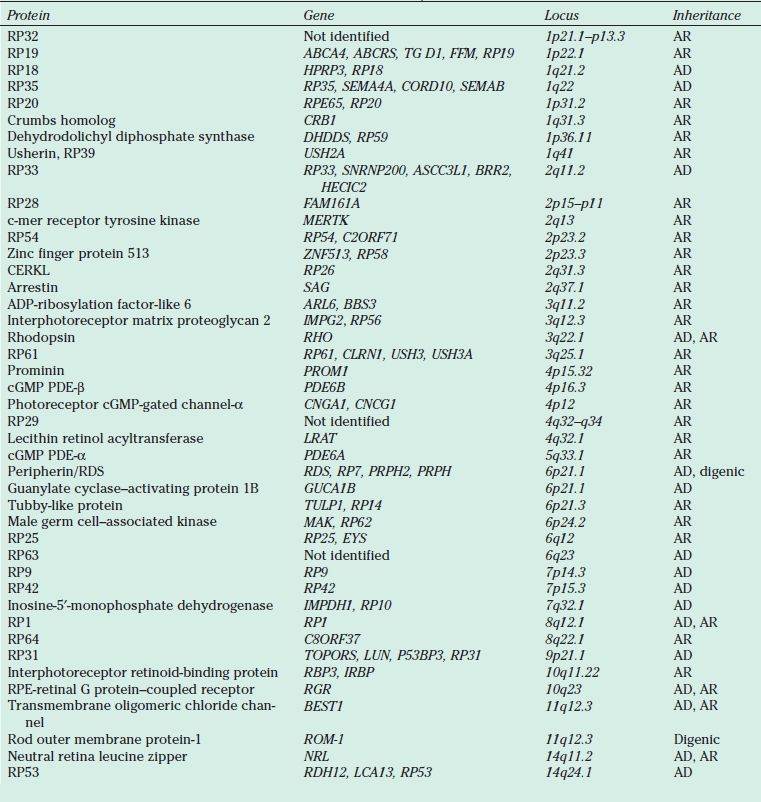
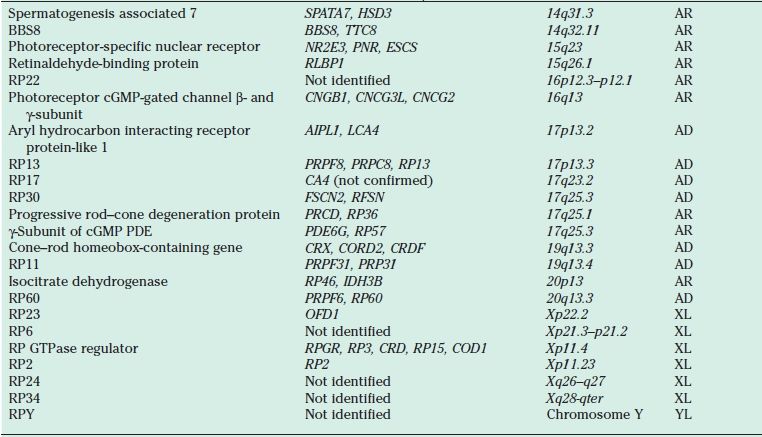
AR, autosomal recessive; AD, autosomal dominant; cGMP, cyclic guanosine monophosphate; PDE, phosphodiesterase; RDS, retinal degeneration slow; RPE, retinal pigment epithelium; XL, X-linked; YL, Y-linked.
Autosomal Recessive Retinitis Pigmentosa
AR RP presents in the first two decades of life and generally has a more fulminant course and poorer prognosis than AD RP. Vision typically deteriorates below 20/200, with significant loss of VF. Field loss can be a more significant contributor to visual dysfunction than acuity and often is first noted 30 to 50 degrees from fixation. These scotomas enlarge, deepen, and coalesce forming a ring of peripheral VF loss, sparing small islands of temporal field. These islands of vision might contribute to some visual function initially, but soon become smaller or not contiguous enough to be useful. Carrier state can be detected by clinical exam and ERG studies. There can be subtle changes in the fundus showing pigment disturbance, migration, and clumping. A golden-metallic tapetal reflex can be seen in some patients in the temporal periphery. ERG will reveal subnormal scotopic and photopic responses. Consanguinity suggests AR RP, if no other form of transmission is suggested by the pedigree.
Many of the genes associated with AR RP are involved with photoreceptor development, the phototransduction cascade, and vitamin A metabolism. Mutations in the catalytic domain of the beta subunit of rod cyclic guanosine monophosphate (cGMP) phosphodiesterase (PDE)-β result in a loss of function of cGMP PDE, leading to accumulation of cGMP levels, which may be toxic to rods (6). Mutations within this protein are the most common of the identified changes in patients with AR RP and account for 4% to 5% of cases. Mutations in the cGMP PDE alpha subunit have also been identified as causing AR RP (7). The cGMP-gated cation channel proteins are heterotetramers of two homologous alpha (CNCG1) and beta (CNGB1) subunits; mutations in these subunits have also been found in AR RP (8,9), (see also Chapter 25).
Recessive RP may be caused by mutations of RPE65, a microsomal protein found exclusively in the RPE, where it accounts for 10% of total membrane proteins (10). Mutations in RPE65 have been reported in Leber congenital amaurosis and recessive cone–rod dystrophy (11,12). Murine studies have shown that although opsin apoprotein is present in the rod outer segment (ROS), no rhodopsin is present. Administration of oral 9-cis-retinal to RPE65-knockout mice resulted in rhodopsin formation with an improvement in ERGs (13). A study using subretinal adeno-associated virus (AAV)–mediated RPE65 transfer in RPE65-deficient canines obtained ERG recordings consistent with restored rod and cone photoreceptor sensitivity 1 to 3 months postinjection. This same study also demonstrated stable ERG responses in RPE65-deficient canines injected 3 years prior. These studies provided the scientific basis for ongoing gene therapy trials (see also Chapter 24).
X-Linked Retinitis Pigmentosa
XL RP usually presents within the first decade of life and is typically the most severe form of RP. These children often present in the first few years of life and can progressively lose VF and acuity over the next decade and show profound impairment of vision (14). XL RP was the first type of RP to be linked to a specific locus by Bhattacharya in 1984 (13). There are now six distinct XL RP loci and three of the responsible genes have been cloned. The RP GTPase regulator gene has been identified in 20% to 30% of XL RP cases (15). This gene is expressed in many tissues and is thought to play a role in membrane transport, possibly involving the shedding and regeneration of rod outer segments (ROS), by regulating retinal GTPase activity. Clinically, affected males show early onset and severe retinal degeneration. Heterozygote female carriers can show mild to no effect to severe expression of the disease, depending on the lyonization of their cells. Rods and cones appear equally affected by ERG in this type of RP (16).
Phenotypic Variation
Considerable variability exists in the RP phenotype due to four factors: (i) locus heterogeneity, in which mutations in different genes are associated with the phenotype; (ii) allelic heterogeneity, in which different mutations in the same gene are associated with the phenotype; (iii) modifier genes that affect phenotypic expression; and (iv) environmental factors that affect phenotypic expression. The RP phenotype can vary in disease stages, penetrance, and expressivity.
Proteins
Proteins important in the pathogenesis of RP may be divided into four categories: (i) photoreceptor structural proteins, (ii) phototransduction cascade proteins, (iii) proteins that regulate photoreceptor and RPE metabolism (including vitamin A metabolism), and (iv) transcription factors.
Pathophysiology
The Visual Cycle
To understand the mutations in RP, familiarity with photoreceptor anatomy, phototransduction pathway, and vitamin A metabolism is necessary (Fig. 21.3). Photoreceptors are light-sensitive cells comprising the outer most layer of the neurosensory retina. They are specialized ciliated neuroepithelial cells that convert light into chemical signals. Their distal parts capture light, and their proximal parts transmit nerve signals. There are two types, rod and cone cells, referring to the cylindrical or cone shape of their photon capturing apical membranes. There are approximately 92 million rods responsible for vision in low light conditions and 5 million cones responsible for day and color vision. Rods are present throughout the fundus, reaching a peak density 20 degrees from the foveola, whereas cones are concentrated in the center of the macula, but can also be found in the rest of the fundus (17). These neuroepithelial cells have a cell body, an inner segment, connecting stalk, and an outer segment. The stalk has the structure of a cilium with nine tubules arising from a basal body in the inner segment and reaching varying heights in the outer segment. All rods contain rhodopsin, the light-sensitive chromophore. Cones contain three opsins sensitive to three different regions of the visual spectrum: blue, green, and red. The ROS is a modified cilium made up of approximately 600 to 1,000 flat discs piled on top of each other. The plasma membrane is extended and flattened into many disks, providing an extensive surface area for capturing incoming photons. At the end of the rims of the disks are lobules. Filaments connect disks to each other and lobules within the same disk (18). After light strikes the apical tips of the outer segments of rods, the outer disks are shed the next morning and phagocytosed by the RPE surrounding the apical portion of the outer segments. New disks are added at the base of the ROS daily.
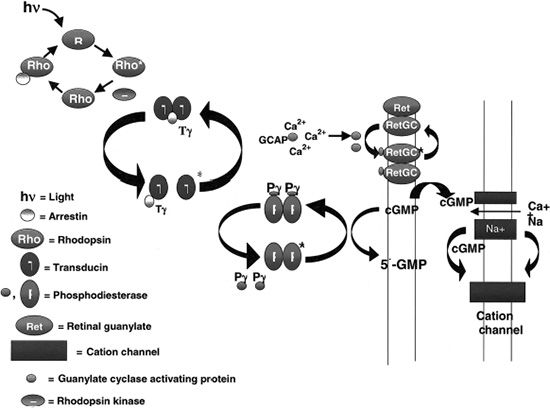
FIGURE 21.3 Phototransduction cascade. (Courtesy of Thomas E. Stout.)
Rhodopsin (RHO) contains seven transmembrane domains and is found on the lamellar side of rod discs (Fig. 21.4). It is the most abundant protein found in rods, constituting up to 85% of the total amount of protein present in the ROS. Rhodopsin is composed of a rod-specific opsin bound to the chromophore 11-cis-retinal. It is important for the initiation of the visual transduction cascade and may be important in ROS structure. Mice lacking a copy of the rho gene have ROSs that are short and disorganized and develop a slow retinal degeneration much like the peripherin/RDS mice (19). In the rim of the ROS, peripherin/RDS and retinal outer segment membrane protein 1 (ROM1) are found as transmembrane proteins. These two proteins are homologous in structure and function, but are expressed in different cells. Peripherin/RDS is expressed in rods and cones, whereas ROM1 is only found in rods. These two proteins form dimers, which interact to form tetramers.
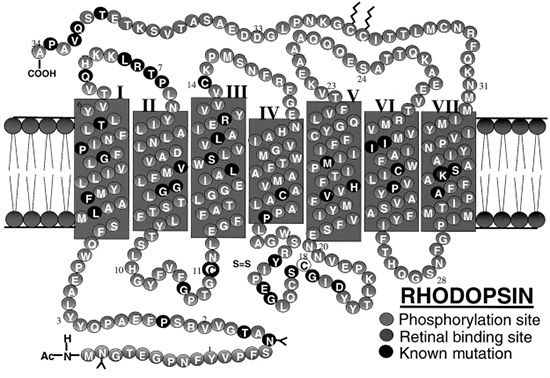
FIGURE 21.4 The complete amino acid sequence of human rhodopsin (RHO) is shown. The protein contains seven transmembrane domains and is found embedded in the bilipid membranes that make up the intracellular photoreceptor discs. (Adapted from Hargrave PA. Rhodopsin structure, function, and topography: the Friedenwald lecture. Invest Ophthalmol Vis Sci 2001;42(1):3–9. Bird AC. Retinal photoreceptor dystrophies LI. Edward Jackson Memorial Lecture. Am J Ophthalmol 1995;119(5):543–562, with permission, www.webvision.med.utah.edu.)
The phototransduction cascade begins when a photon strikes an ROS and is absorbed by 11-cis-retinal transforming it into all-trans-retinal (Fig. 21.3). This event induces a conformational change in rhodopsin, which catalyzes the activation of the G-protein transducin. Many molecules of transducin are activated by a single rhodopsin, starting this signal amplification cascade. Transducin activates cGMP PDE to hydrolyze cGMP. The resulting decrease in cGMP concentration closes the cGMP-gated cation channels in the cell membrane, which results in the hyperpolarization of the cell, slowing the neurotransmitter release at the synaptic terminal (20). Deactivation of RHO starts with phosphorylation by rhodopsin kinase and capture by arrestin. The binding of arrestin to RHO prevents further activation of transducin and releases 11-trans-retinal from rhodopsin (21). The regeneration of the 11-cis-retinal from the all-trans-retinal is mediated by the interphotoreceptor retinoid-binding protein (IRBP), which transports it from the ROS to the RPE cell. Here it is transferred to the cellular retinoid-binding protein and is converted to 11-cis-retinal by RPE-specific isomerase. Lastly, 11-cis-retinal binds cellular retinaldehyde-binding protein (CRALBP) and is oxidized to 11-cis-retinal, which is released from the RPE cells. It is then transported to the ROS by IRBP, where 11-cis-retinal binds rhodopsin all over again. The RPE-specific protein and RPE65 protein, which are involved in this regeneration process, constitute perhaps as much as 10% of the total amount of protein in the RPE cells (22).
The daily shedding of photoreceptor outer segments and renewal of the 11-cis-retinal place a burden on the multifunctional RPE cells. Any malfunction can lead to an accumulation of shed outer segments, all-trans-retinal, and deposits between the RPE and photoreceptor layers.
Genetics
Rhodopsin
This light-activated protein, part of the G-protein–coupled receptor superfamily, consists of the apoprotein opsin with 348 amino acid residues and the chromophore 11-cis-retinal covalently bound to Lys-296 in the seventh transmembrane helix via a protonated Schiff base (PSB) linkage. The rhodopsin gene, located on chromosome 3q, was the first in which mutations associated with RP were identified. Over 100 different mutations in the opsin gene have been described since the initial p.Pro23His mutation in the intradiscal domain of the protein was reported in 1990. Almost all of these mutations result in the synthesis of an abnormal protein due to a single amino acid substitution (23). Mutations in the rhodopsin gene account for 25% of all AD RP in the United Kingdom, Europe, and the United States (24–26). The p.Pro23His mutation is the only one to have a sizable number of cases. It has been detected in 12% of the RP population in the United States, although it has not yet been found in any of the European cases (27). A new mutation (p.Pro23Ala) has been recently reported. It is much less frequent than the p.Pro23His mutation and causes a more benign form of AD RP, with greater preservation of ERG amplitudes at presentation and a milder course (28). Most mutations have been found only in a small number of patients and are associated with AD RP. Misfolding of the rhodopsin protein has been proposed as one of the main biochemical consequences resulting from mutations in the rhodopsin gene (29). Molecular chaperones have also been implicated as a source of dysfunction (30). The partial or complete misfolding of rhodopsin can result in the inability of the mutant protein to bind 11-cis-retinal. Other rhodopsin mutations include p.Thr58Arg and p.Gly106Asp. These two mutations produce a sectoral AD RP, characterized by loss of the superior VF with a corresponding inferior retinal degeneration (31). Rare cases of AR RP have been reported, including a proband from a consanguineous marriage who was homozygous for the p.Glu249Ter mutation. This mutation causes premature termination and would be expected to result in complete absence of functional protein. The heterozygous parents and siblings did not have RP, but abnormalities in rod photoreceptor function were noted (32,33).
Three rhodopsin mutations that cause both AD CSNB and AR RP have been reported: p.Gly90Asp, p.Thr94Ile, and p.Ala292Glu are located in the region of the protein that is involved with the PSB linkage to 11-cis-retinal (34,35). The resulting phenotype is a stationary retinal dystrophy. These mutations do not seem to cause the gross structural misfolding that is predicted for other rhodopsin mutations (36), which may explain the more benign phenotype.
Functional and expression studies of mutations in rhodopsin in vitro have defined three classes of mutants based on the synthesis of rhodopsin, regeneration of 11-cis-retinal, and the glycosylation pattern (37,38). Class I mutants (15%) cluster in the first transmem-brane region toward the C-terminus end of the molecule. These molecules resembled the wild type in terms of yield, regeneration with 11-cis-retinal, and subcellular location. Class II mutants (85%) were unstable or folded improperly, accumulating in the rough endoplasmic reticulum. Although cell-culture studies showed two classes of mutants, no direct correlation was observed between classes and the phenotype of the patients with either class of mutation. This study showed a different mechanism than had been hypothesized in earlier studies that speculated a constitutive activation of the phototransduction cascade. Animal studies have shown that continuous exposure to light leads to retinal degeneration (39). Examination of a constitutively activated codon 296 mutation at the binding site of 11-cis-retinal in humans, which might turn on the cascade continuously, actually revealed in a mouse model that it was the continuous shutoff of the visual cascade that triggered the degeneration of the photoreceptors (40).
As mentioned earlier, rhodopsin is a major constituent of ROS, accounting for 85% of the protein content. It has an important structural role in the ROS. Generation of mice with a targeted disruption of the rhodopsin gene showed that homozygous knockouts did not have ROS and they lost their photoreceptors within 3 months. Heterozygotes, on the other hand, retained most of their photoreceptors, but they developed short, stubby ROS and underwent inexorable retinal degeneration as they got older, much like that seen in peripherin-deficient/RDS mice (19).
The abundance of rhodopsin, its importance for structure of the ROS, and the findings from animal studies suggest that the accumulation of most forms of mutant rhodopsin in subcellular structures may cause disruption of cellular processes necessary for the proper renewal and shedding of ROS and thus result in photoreceptor cell death.
Peripherin/RDS and ROM1
Peripherin is a transmembrane protein abundant in the outer disk rims in both rods and cones, while ROM1 is a transmembrane protein abundant only in rods. They are structural proteins of the outer segment and are estimated to be abnormal in 5% of all AD RP cases (41). The genes that encode peripherin/RDS and ROM1 are located on chromosome 6 and chromosome 11, respectively. These proteins play an important role in maintaining disk curvature and possibly anchoring these disks to the plasma membrane (42). More than 40 different mutations in the human peripherin/RDS gene have been identified. These are usually inherited in an AD fashion. Studies of rds mice have demonstrated that heterozygous mice with a 50% reduction of peripherin/RDS develop short and stubby photoreceptors that slowly degenerate (43). Homozygous mice do not develop any outer segments. Mutations in this gene in humans often result in a slow retinal degeneration, with a delayed onset. There is a slow dysfunction of both the rod and cone photoreceptors with the appearance of yellow deposits in the RPE. A variety of phenotypes have been observed within the same family, including RP, pattern dystrophy (Fig. 21.5), fundus flavimaculatus (Fig. 21.6), and retinitis punctata albescens (44–46). Transgenic mice with specific mutations in both copies of the peripherin/RDS gene have outer segment dysplasia and accelerated photoreceptor cell death compared with heterozygous mice (47).
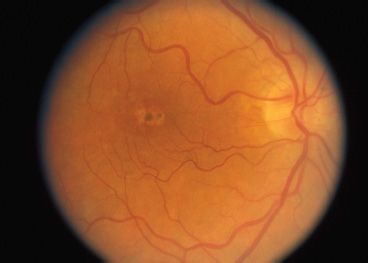
FIGURE 21.5 Autosomal recessive pattern dystrophy.
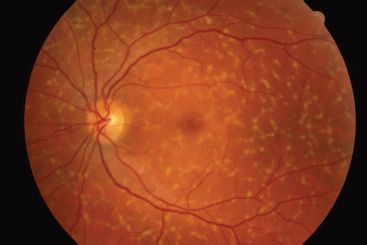
FIGURE 21.6 Fundus flavimaculatus. (Photograph courtesy of Richard Weleber, MD.)
The relationship between peripherin/RDS and ROM1 was elucidated by families in which members with RP were heterozygous for the p.Leu185Pro mutation in the peripherin/RDS gene and a mutation in the ROM1 gene, suggesting a digenic pattern of inheritance. Those who were heterozygous for only one of these two genes did not have the phenotype (48).
Differential Diagnosis
The diagnosis of RP is made by the clinical presentation of nyctalopia, fundus findings, loss of peripheral VF, family history and examination, and ERG testing. RP does not present in the 1st year of life. If an infant presents with wandering eyes from birth and no visual behavior, oculodigital reflex, or paradoxical pupillary response to light, other diagnoses should be considered including Leber congenital amaurosis. Genetic testing (genomic DNA sequence analysis) for the diagnosis of RP is becoming more common. Syndromic causes should be excluded by careful history and physical examination. An increased incidence of optic nerve head drusen (Fig. 21.7), pigmented vitreous cells (due to RPE migration), cystoid macular edema (CME), posterior sub-capsular cataract, and epiretinal membranes may also provide clues to the diagnosis.
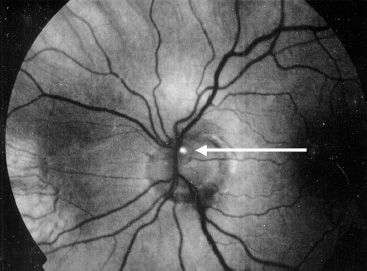
FIGURE 21.7 Red-free photograph of optic disc drusen demonstrating autofluorescence (arrow) in autosomal recessive retinitis pigmentosa.
Diagnostic Studies
ERG testing is useful, even in the 1st year of life. Testing done before 6 months of age should be repeated after 6 months, as visual maturation may not have been complete at the initial testing. Standard ERG testing is performed according to the International Society for Clinical Electrophysiology of Vision guidelines (see also Chapter 10). Tested responses include (i) a bright white flash, which elicits a mass response of combined cone and rod function; (ii) a scotopic response of rods after 45 minutes of dark adaptation; and (iii) a photopic response and a 30-Hz flicker, which measures the cone function. RP may cause a reduction in any of these responses. The implicit time may also be delayed, which measures the onset of the photopic b wave after a flash of white light. Pattern ERG testing may help evaluate macular function.
The electrooculogram measures the standing potential of the RPE and may be abnormal, showing an absence of the light rise. Dark adaptometry measures quantitatively the kinetics of cone and rod recovery following a bright flash and can be useful to confirm RP in difficult cases. The rod–cone break will be abnormal. The confocal scanning laser ophthalmoscope can image the autofluorescence of the fundus, which results mostly from the RPE. Increased autofluorescence can indicate increased RPE dysfunction, as a result of overloading of these RPE cells in areas of excessive shedding of photoreceptor outer segments (49).
Fluorescein angiography, often not necessary for diagnosis can reveal hypofluorescence from pigment clumping or hyperfluorescence from RPE window defects and leakage from CME, which can be responsible for potentially reversible loss of vision in RP patients.
Management
Although no clearly effective therapy is currently available for patients with RP, patients should be periodically monitored for disease progression and the possible development of reversible causes of vision loss. All patients suspected to have RP should be periodically assessed by kinetic perimetry. In addition to providing useful information about disease progression, serial perimetry has practical influences on driving ability, blind registration, and eligibility for disability funding. Serial ERGs can be followed if the diagnosis is suspected in the first few years of life. RP will result in a diminution of the scotopic and combined responses. Consultation with a low-vision specialist for optical devices that optimize contrast, increase magnification, or provide a dark background for reading text may be helpful. Patients with decreased central vision should be evaluated for CME that may be responsive to systemic or topical carbonic anhydrase inhibitors. Because cataracts may reduce vision, cataract extraction with intraocular lens placement may improve central visual acuity in select cases. Vision loss may also accompany a Coats-like exudative process with retinal detachment that may respond to laser or cryoablative therapy (Fig. 21.8). Limiting light exposure might be bene-fcial to some patients, and sunglasses may decrease light sensitivity. Although controversial, many physicians treat RP patients with 15,000 IU of oral vitamin A per day (50). Somatic gene transfer may play a therapeutic role in the future as many genes responsible for RP are cloned and methods for gene transfer into terminally differentiated retinal and RPE cells improve. Researchers in Germany recently used archaebacterial halorhodopsin expression via AAV to reestablish the phototransduction cascade and light sensitivity in Cnga3-/- and Rho-/- mice (51). In vitro fertilization with zygote screening may also be an option for parents who plan to have additional children.
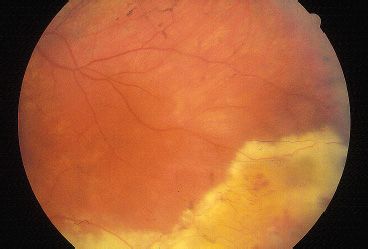
FIGURE 21.8 RP with Coats-like exudative peripheral retinal detachment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


