FIGURE 20.1 Leber optic neuropathy disc in a patient harboring the 11778 mtDNA mutation demonstrating circumpapillary telangiectasia and swelling of the nerve fiber layer around the disc. (Courtesy of N. R. Miller.)
Management
No established treatments are available at the present time to prevent vision loss, arrest the progression of vision loss, or restore vision. However, significant progress has been obtained in recent years toward the development of treatment for LHON. A recent open-label clinical trial on five LHON subjects suggested that EPI-743, an orally absorbed small molecule, which targets the enzyme NADPH quinone oxidoreductase 1, may be beneficial in arresting visual loss in LHON (49,50). In addition, the results of a randomized placebo-controlled trial of idebenone (a well-tolerated, short-chain coenzyme Q analog) have recently been reported. Although the end point was not statistically significant, post hoc analysis showed benefits in some patients, and the authors concluded that idebenone appears to ameliorate the visual outcome particularly among patients with the m.11778G>A and m.3460G>A mutations (51,52).
Another potential future treatment technique for LHON is gene therapy. A gene-replacement technique in cell culture has been described in which a defect in complex I function in cells harboring the 11778 mutation was restored (53). The in vivo feasibility, efficacy, and safety of such a technique are currently being evaluated (54,55).
AUTOSOMAL DOMINANT OPTIC ATROPHY
Prevalence
ADOA, also known as Kjer disease, is one of the most common forms of autosomal inherited optic neuropathies, affecting 1:35,000 (Northern England) to 1:12,000 (Denmark) based on clinical and molecular data. The penetrance may be as low as 40% (5,6,56,57).
Genetics and Pathophysiology
Approximately 60% of ADOA patients harbor mutations in the OPA1 gene, whereas only a few cases of OPA3 mutations have been reported. The OPA1 gene consists of 29 exons spread over 100 kb of genomic DNA, and alternative splicing results in eight isoforms. OPA1 encodes a dynamin-like mitochondrial GTPase, and many of the over 200 pathogenic mutations reported cluster in the GTPase domain and the dynamin central region. Single base pair mutations are the most common, followed by deletions and insertions. OPA1 testing by sequencing is useful, but some large-scale rearrangements may be missed by this technique (5,7,58,57,59) (eOPA1 database [http://lbbma.univ-angers.fr/lbbma.php?id=9]).
The OPA1 protein associates with mitochondrial IM phospholipids such as cardiolipin and plays a pivotal role in the maintenance of mitochondrial morphology. A recent study in an OPA1 transgenic mouse model demonstrated that mitochondrial fragmentation precedes dendritic loss, thereby directly linking retinal ganglion cell (RGC) synaptic architecture to OPA1 function (60).
Interestingly, studies suggest that patients with ADOA are born with fewer optic nerve axons and smaller optic discs, supporting the hypothesis that subsequent visual loss depends on further age-related loss of fibers, which also occurs in controls. It may also suggest OPA1 involvement in eye development (61).
The pathomechanism of OPA3 mutations associated with the autosomal recessive disorder, 3-methylglutaconic aciduria type III (Costeff syndrome), seems to be similar to that of OPA1 mutations, as mutant OPA3 mice also showed disrupted mitochondrial morphology in the retina confirming the importance of the mitochondrial network in retinal function (59,62). Notably, other diseases linked to abnormal mitochondrial morphology, such as Charcot-Marie-Tooth type 2A and hereditary motor and sensory neuropathy type VI, also exhibit ocular manifestations (52).
Ocular Manifestations
The classical presentation of autosomal dominant optic neuropathy includes insidious childhood onset with selective loss of RGCs resulting in optic nerve degeneration. The phenotype is generally milder than LHON, but the expression is variable, also within families, ranging from subclinical manifestations to legal blindness and mean visual acuities of 6/18 to 6/60 (63). Patients may show generalized dyschromatopsia, and isolated tritanopia is uncommon. Visual field defects in ADOA reflect the tendency of the disease for involvement of the papillomacular bundle, manifesting central, cecocentral, or paracentral scotomata with preserved peripheral fields. Similar to LHON, the pupil response to light is relatively preserved (5,60). Potentially, OPA1 variants are also associated with the risk for developing normal tension glaucoma in adulthood (64).
Systemic Manifestations
ADOA is characterized by the preferential loss of RGCs. However, OPA1 is expressed also in the inner ear, and the most frequent extraocular manifestations of OPA1 mutations are sensorineural deafness present in about two-thirds of the cases (ADOAD). Additionally, about 20% of patients with OPA1 mutations will develop a more severe disease variant (ADOA+), with additional neuromuscular features including ataxia, myopathy, peripheral neuropathy, CPEO, or multiple sclerosis–like disorders. Muscle pathology resembles classical mitochondrial diseases with cytochrome c oxidase–deficient fibers and ragged red fibers. Patients also harbor mtDNA deletions in affected muscle tissue linking OPA1 with mtDNA stability. Notably, individuals with mutations that affect the OPA1 GTPase domain are more likely to develop multisystem neurologic disease (5,6,58,64).
Differential Diagnosis
LHON; other inherited optic neuropathies such as Leigh syndrome, Wolfram syndrome, and Costeff optic atrophy; and toxic neuropathies may be differentiated from ADOA by clinical exam, systemic manifestations, and family history.
Management
Current management includes genetic counseling. Future treatment may include antioxidants and, potentially, gene therapy (65).
KEARNS-SAYRE SYNDROME AND CHRONIC PROGRESSIVE EXTERNAL OPHTHALMOPLEGIA
Genetics and Pathogenesis
A mitochondrial defect was suspected to underlie the KSS based on the inheritance pattern observed in several affected families (66). Indeed, it was later found that both KSS and CPEO are associated with mtDNA deletions (11,67). The deletions typically are 1.3 to 7.6 kb in length, but there is an increased prevalence of the 4.9-kb deletion. Interestingly, identical mtDNA deletions can lead to CPEO or KSS (with its additional systemic manifestations), and this may at least in part depend on the distribution of mtDNA deletions in different tissues.
PEO with multiple mtDNA deletions is also associated with mutations in the nuclear genome and can be transmitted in autosomal dominant or autosomal recessive modes of inheritance. Three genes are associated with autosomal dominant PEO: adenine nucleotide translocator-1 (ANT1), which is located on chromosome 4 and encodes a muscle-specific adenine nucleotide translocator; Twinkle (PEO1, C10orf2), encoding the mitochondrial helicase; and mitochondrial polymerase gamma (POLG), which is located on chromosome 15 and encodes the alpha subunit of polymerase gamma. Mutations in POLG are the most frequent cause of all forms of familial PEO. Approximately 150 POLG mutations have been associated with a wide range of mitochondrial diseases presenting with a continuous spectrum of overlapping symptoms and signs, ranging from fatal infantile disease to PEO presenting in adulthood (68,69) (http://tools.niehs.nih.gov/polg/). All three genes are associated with mtDNA replication and maintenance; thus, alterations in their function might lead to mtDNA deletions (70). Genetic analysis in these patients usually finds multiple concurrent mitochondrial mutations, suggesting that the nuclear DNA mutations induce instability of mtDNA replication (14,71). Notably, external ophthalmoplegia and other ocular manifestations are also found in the autosomal recessive mtDNA depletion syndrome, mitochondrial neurogastrointestinal encephalomyopathy (MNGIE), which is associated with mutations in the TK2 gene on chromosome 16. The TK2 protein is a mitochondrial deoxyribonucleoside kinase that phosphorylates thymidine, deoxycytidine, and deoxyuridine (13).
Ocular Manifestations
KSS is characterized by the triad of external ophthalmoplegia (CPEO), pigmentary retinopathy, and heart block (9,72). Onset is in the first or second decade of life. CPEO is characterized by a bilateral symmetric ptosis associated with ophthalmoplegia and orbicularis oculi weakness. In addition, KSS patients manifest an atypical pigmentary retinopathy in which the macula is often the first part of the retina to be affected followed by the retina periphery. In some patients, all parts of the retina are affected simultaneously. Histology of a retina from KSS patients showed atrophy of the RPE and outer retina that was most marked posteriorly (10). In addition to the different retinal involvement pattern, KSS also differs from classic RP in that there is usually no blood vessel attenuation and no waxy optic nerve appearance accompanying the pigmentary retinopathy. Furthermore, visual acuity, visual fields, and electroretinogram (ERG) are usually only mildly affected.
Systemic Manifestations
Neurologic manifestations in KSS may include cerebellar ataxia, hearing loss, dementia, and weakness of facial, pharyngeal, trunk, and extremity muscles. Heart block is a characteristic finding (9,72,73). POLG mutations have an overlapping spectrum of symptoms involving the nervous system as well as liver and muscle. One of the most severe manifestations is fatal hepatic failure, the Alpers-Huttenlocher syndrome, which is caused by over 50 different POLG mutations. Other mtDNA deletion syndromes may also present with extraocular manifestations including hearing loss, muscle weakness, and neuropathy (69). COX-deficient muscle fibers, a hallmark of mitochondrial disorders, are frequently present, and MNGIE is characterized by prominent gastrointestinal symptoms (13,68).
Diagnosis
Diagnosis is based on the typical ocular, neurologic, and cardiac manifestations. Lumbar puncture can demonstrate an increased cerebrospinal fluid protein level (the underlying cause of which is still unknown). Electrocardiography is useful in identifying heart block, and genetic testing can establish the diagnosis in cases of doubt. The identification of a diseasecausing mutation confirms the diagnosis and facilitates genetic counseling.
Management
While there is no cure, careful correction of ptosis or strabismus may be beneficial in selected cases. The cardiac conduction defect may be life threatening and is treated with a pacemaker.
Tetracycline treatment has been evaluated in PEO; however, the results did not formally support its effectiveness (74). At present, only MNGIE has been successfully treated by allogeneic hematopoietic stem cell transplantation (75). Genetic therapies are being developed for KSS (76).
NEUROPATHY ATAXIA RETINITIS PIGMENTOSA AND LEIGH SYNDROMES
NARP and Leigh syndromes are two inherited mitochondrial diseases associated with the same mtDNA mutation at nucleotide position 8993. Leigh syndrome is also associated with other mtDNA mutations (http://www.mitomap.org/bin/view.pl/MITOMAP/ClinicalPhenotypesPolypeptide) and mutations in several nuclear genes.
Genetics and Systemic Manifestations
The T>G point mutation at nucleotide 8993 of mtDNA causes a substitution of a highly conserved leucine by an arginine residue in the ATPase 6 protein (19). mtDNA with both the T and the G is present in the same subject (heteroplasmy), but the proportion of the mutant mtDNA differs among patients. Usually, a high proportion of mutant mtDNA is associated with more severe clinical manifestations (3,77,78). Two clinical syndromes were described in association with the 8993 mtDNA mutation. The first, subacute necrotizing encephalomyopathy or Leigh syndrome, is a severe disease, with patients showing developmental delay, ataxia, psychomotor regression, seizures, peripheral neuropathy, and optic atrophy. Onset is usually during infancy with progression to death within months to years. The 8993 mutation was found to be a common cause of Leigh syndrome, and most of the patients had more than 90% of mutant mtDNA (77). The second entity, NARP syndrome, first reported by Holt et al. in 1990 (19), is usually milder than Leigh syndrome. Patients show a variety of clinical manifestations such as migraine, sensory neuropathy, proximal muscle weakness, ataxia, seizures, dementia, and pigmentary retinopathy (3,19,22). Patients with the NARP syndrome usually carry around 80% of mutant mtDNA. At levels below 75%, carriers may show pigmentary retinopathy, suffer from migraines, or be totally asymptomatic (3,78). Leigh disease has also been associated with mutations in other mtDNA genes mostly in ND3 and ND5 (79,80).
In some patients with Leigh syndrome, mutations have also been found in nuclear genes, including SURF1 and other cytochrome oxidase assembly genes and POLG (81,82). However, mutations have not been identified in about half of Leigh syndrome patients even after extensive molecular testing. Notably, age of onset, clinical symptoms, and prognosis are similar in Leigh syndrome patients with mtDNA or nuclear DNA mutations (80).
Ocular Manifestations
The ocular abnormalities in NARP syndrome were reported in most cases as typical RP or as pigmentary retinopathy (3,19,78) (Fig. 20.2). Puddu et al. (83) described three patients with NARP syndrome, two of whom had ocular manifestations. The fundus examination showed bone spicule pigmentation in the midperiphery, and despite being described as RP, the ERG findings were normal except for subnormal photopic responses. In another report, Ortiz et al. (22) described eight patients from two families with the 8993 mutation in whom the ocular manifestations ranged from a mild salt-and-pepper retinopathy to severe RP-like changes with maculopathy. Four of these patients underwent ERG examination. One showed normal results, and three revealed rod–cone-type dysfunction. In five patients who had perimetry, four had normal visual fields and one had a paracentral scotoma.
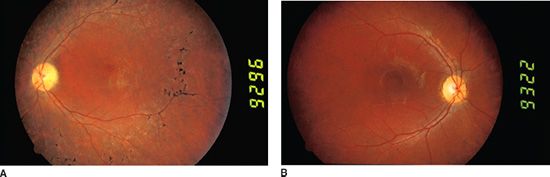
FIGURE 20.2 Color fundus photographs of two NARP patients demonstrating the variable ocular manifestation of the disease. A: A 16-year-old male NARP patient with blots of pigment along with multiple small retinal scars, narrowing of the blood vessels, and pigmentary changes in the fovea. The optic disc appears normal. This patient had reduced visual acuity, had nyctalopia, and showed bull’s-eye maculopathy on fluorescein angiogram. The visual field of this eye had central and paracentral scotomata, and ERG showed decreased cone-and rod-derived amplitudes. B: The sister of the patient presented in panel A was 12 years old at that time. She manifested a bull’s-eye maculopathy with temporal optic disc pallor. No pigmentary changes were apparent. This patient had mild reduction of visual acuity and decreased cone-derived (but not rod-derived) ERG responses. This patient also had episodic ataxia and mild mental retardation.
Chowers et al. (4) described three members from a family harboring the 8993 mtDNA mutation in whom great variability in the clinical manifestations of the disease was observed. Two of the three patients had ocular abnormalities that were different from classic RP. The clinical findings, visual fields, and ERG were typical of cone–rod dystrophy in one of the patients and progressive cone dystrophy in another patient. Other retinal findings in NARP include a bull’s-eye maculopathy and rod–cone type of retinal dystrophy (16).
As discussed, there is great variability in the ocular and systemic manifestations among patients carrying the mtDNA 8993 mutation. Several factors could account for the spectrum of abnormalities observed. First, the burden of mutant mtDNA in a particular tissue may vary thus influencing phenotype, with a higher proportion of mutant mtDNA in blood lymphocytes being a marker for more severe clinical manifestations. However, Chowers et al. (4) observed that patients with a lower percentage of mutant mtDNA in the blood lymphocytes can have a more severe ocular disease compared with patients with higher proportion of mutant mtDNA. This can be partially explained if the proportion of mutant mtDNA in the retinas differs from that in the blood or other tissues, especially the central nervous system (CNS), but such differences have not yet been demonstrated (78).Differences among patients may also depend on age. Ortiz et al. (22) described two patients, aged 12 and 14 years, who suffered from NARP syndrome and had a rod–cone type of ERG dysfunction. The retinopathy of these patients was much less advanced than that of patients reported by Chowers et al. who were of a similar age but with a predominantly cone dysfunction. Thus, patients with the NARP syndrome may manifest either rod- or cone-dominant retinal dystrophy at a similar age. It is likely that other factors, not yet understood, modulate the phenotypic expression of the 8993 mtDNA mutation. In support of this, two of the three reported NARP pedigrees undergoing ERG had predominantly cone dysfunction, while the third pedigree had predominantly rod dysfunction. This finding could not be explained by the differences in age, stages of the disease, or percentage of mutant mtDNA among families.
Diagnosis
Diagnosis is based on identification of the typical combination of ocular and systemic manifestations along with the mitochondrial inheritance pattern. Ancillary tests may include ERG, color vision, and fluorescein angiography. Definitive diagnosis is made by molecular testing.
Management
No treatment has been shown to be of benefit in arresting the progression of retinal dystrophy in patients carrying the mtDNA 8993 mutation. In addition, patients should be referred to a clinical geneticist for evaluation of medical and family history because although the 8993 mutation is transmitted from mother to each of her offspring, the proportion of mutant mtDNA in each offspring can differ considerably from that of the mother and with it the clinical manifestations. Further research is required to evaluate if novel therapies for mitochondrial disorders such as EPI-743 and idebenone or gene therapy may be beneficial for NARP and Leigh syndrome patients (50).
MITOCHONDRIAL ENCEPHALOMYOPATHY, LACTIC ACIDOSIS, AND STROKE-LIKE EPISODES SYNDROME
While vision loss in mitochondrial genetic disorders more commonly occurs from retinal degeneration or optic nerve disease, vision loss can occur from damage to the retrochiasmal visual pathways as in mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) (84,85
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


