FIGURE 19.1 Scanning electron micrograph of an RPE monolayer from a patient with STGD1/FF. Note the marked heterogeneity of the RPE cells engorged with lipofuscin. Aggregates of engorged cells may correspond to flecks identified on clinical examination. (Courtesy of Ralph C. Eagle Jr., MD, Department of Pathology, Wills Eye Hospital, Philadelphia, PA.)

FIGURE 19.2 Histophotomicrograph of an intact monolayer of RPE from a patient with STGD1/FF. The enlarged RPE cells demonstrate abundant periodic acid-Schiff–positive material in the apical compartment, consistent with lipofuscin-like material. (Periodic acid-Schiff stain, original magnification ×250, Courtesy of Ralph C. Eagle Jr., MD, Department of Pathology, Wills Eye Hospital, Philadelphia, PA.)
An extensive review of RPE cellular structure and function is presented in The Retinal Pigment Epithelium: Function and Disease (33). Briefly, the apical surface of the RPE cell extends microvilli to encompass the rod and cone outer segments. The apex of the RPE cell is responsible for the phagocytosis of shed photoreceptor outer segment discs forming phagosomes in the apical RPE cell cytoplasm. The eventual fate of the phagosome is to be incorporated into the lysosomal system for degradation and partial recycling. Cathepsin D is an important protease involved in digestion of the rhodopsin-rich disk membranes. Perturbation of cathepsin D, or other catabolic enzyme systems such as ubiquitination, can lead to the buildup of intracellular and extracellular debris (34).
Lipofuscin normally accumulates in the RPE in an apparent biphasic pattern with one peak occurring between 10 and 20 years of age and the second peak occurring around 50 years of age (35). Lipofuscin buildup is greatest in the posterior pole, especially in the macula, but sparing the fovea (36). Macular pigments composed of lutein and zeaxanthin may influence the accumulation of lipofuscin in the fovea (37). Moreover, specialized cones that reside in the fovea may have cellular membrane properties or different visual pigments that preclude the formation of lipofuscin in this region.
Lipofuscin is contained within granules of relatively uniform size. It is a lipid–protein aggregate that autofluoresces when excited by short wavelength light. The composition of RPE lipofuscin is controversial, but most believe that partially degraded outer segment disks and autophagy processes contribute to the bulk of lipofuscin (38). There is a growing body of evidence that lipofuscin may actually induce oxidative damage by acting as a photosensitizing agent generating reactive oxygen species (39). As a design of function, the macula is exposed to a lifetime of light irradiation, including blue light wavelengths that have been shown to induce reactive oxygen intermediates (40). In addition, biochemical properties of lipofuscin may actually interfere with the enzymatic pathways of degradation by influencing lysosomal pH (41). Thus, as lipofuscin accumulates, the cellular catabolic machinery may be damaged or inhibited leading to more accumulation. This cycle would theoretically continue throughout life until the RPE cell is overwhelmed and cellular death and atrophy occur leading to subsequent overlying photoreceptor cell death.
The ABCA4 gene encodes a protein that is a member of a well-characterized superfamily of proteins known as the ABC transporters. ABCA4 is believed to encode a protein that is involved in the transport of all-trans retinal from the photoreceptors to the RPE. Using knockout mice (abcr-/-), studies have suggested that ABCA4 functions as an export flippase for retinyl derivatives (42). This transport reaction is part of a larger cycle that replenishes the supply of the critical light-sensing form of vitamin A, 11-cis retinal, in the photoreceptor outer segments. The process of light absorption in the photoreceptors converts 11-cis retinal to a derivative called all-trans retinal, which is released into the disk membrane and then chemically modified and transported to an RPE cell. Defects in the ABCA4 protein, however, slow the chemical modification of all-trans retinal, resulting in a buildup of this compound. Unfortunately, all-trans retinal spontaneously reacts at a low rate with membrane lipids to form a substance known as A2E. Sparrow et al. (43) have continued to provide evidence that A2E impairs RPE viability by sensitizing the cells to light damage.
Earlier work suggested that ABCA4 produced a retina-specific transporter protein exclusively confined to the disk membrane of rod outer segments (44). Yet clinically, STGD1/FF can severely impact central macular function, and some patients experience symptoms associated with cone function. In the foveal region, photoreceptor density approximates 200,000 cells/mm2. These densely packed neuronal cells are primarily cones but have specialized architecture that resembles rods (45). Molday et al. (46) have recently suggested that ABCA4 protein is present in both rod and cone photoreceptors. They conclude that the loss in central vision experienced by many STGD1/FF patients arises directly from ABCA4-mediated foveal cone degeneration. Elegant work by Scholl et al. (47) has shown altered electrophysiology in both the rod and cone systems in patients with genetically confirmed STGD1/FF.
The spectrum of disease that can occur even in the same family is broad. Usually for a recessive disease, obligate carriers of a mutant allele appear to be completely unaffected by the disease traits. Sensitive modern equipment, however, can sometimes detect subclinical alterations in function that are suggestive of the disease. The term variable expressivity is used when the severity of the phenotype differs among individuals with identical mutations. As mentioned earlier, the “classic” STGD1/FF patient has a bull’s-eye maculopathy with a beatenbronze appearance. In most cases, small yellow-white flecks with a pisciform shape surround the atrophic area and can be separated from the central lesion by a zone of apparently unaffected RPE (Figs. 19.3 and 19.4).
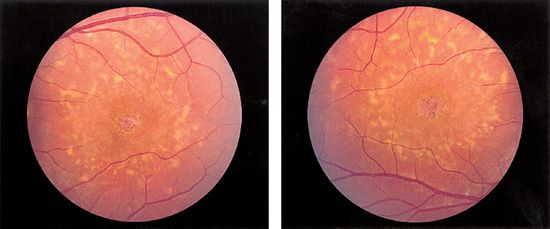
FIGURE 19.3 Left and right:Color fundus photographs of a typical STGD1/FF patient with pisciform yellow-white flecks and a central beaten-bronze bull’s-eye maculopathy. There usually is symmetry in clinical appearance when comparing eyes within an individual. (Courtesy of James Gilman, CRA, Ophthalmic Photography, Department of Ophthalmology, Moran Eye Center, Salt Lake City, UT.)
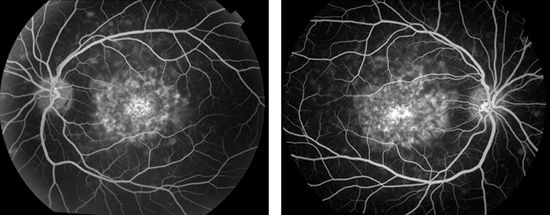
FIGURE 19.4 Left and Right: FA of the same patient as shown in Figure 19.3 shows central areas of hyperfluorescence consistent with window defects, indicating pigment epithelial atrophy. Note the lack of hyperfluorescence corresponding to the flecks. Very late in an angiogram, these flecks may stain. (Courtesy of James Gilman, CRA, Ophthalmic Photography, Department of Ophthalmology, Moran Eye Center, Salt Lake City, UT.)
Following patients with macular flecks using fundus autofluorescence (FAF) imaging has provided further understanding of the temporal evolution of flecks and RPE changes (32). A single fleck, followed with FAF imaging, is initially hyperautofluorescent. Subsequently, that fleck will progressively lose autofluorescence and eventually become hypoautofluorescent. This sequence of change in autofluorescence signifies an early engorgement of RPE cells with lipofuscin and its related components. As burdened RPE cells begin to atrophy, they become gradually less hyperautofluorescent until subsequently containing less lipofuscin than adjacent retina and RPE and appearing hypoautofluorescent on FAF imaging. Furthermore, the location of flecks changes in a nonrandom manner over time. Flecks appear to progress in a radial manner, beginning in the central macula and extending outward in all directions. As new flecks appear toward the peripheral macula, they leave behind an enlarging area of RPE and photoreceptor atrophy.
In patients with STGD1/FF, the age of disease onset and the age of presentation can vary slightly because patients often present only after central visual deterioration has impacted their lifestyle or ability to function. A hallmark of presentation is, however, that the visual function was at one time entirely normal. Children with STGD1/FF have reported visual complaints very early in life, as early as 3 years of age. The mean age of symptomatic visual impairment in patients with central macular involvement appears to be in the early to midteens. With specific questioning, many of these young patients will report visual symptoms that began several years prior to presentation. This hindsight reporting suggests that children may have delayed diagnosis that could impact future interventions.
At presentation, there is a general symmetry of the fellow eyes in regard to function and clinical findings; however, there are always exceptions. In the initial stage, there may be no clinical findings associated with visual disturbance. Of the 56 patients followed by Aaberg Sr., all but 1 patient demonstrated symmetric ophthalmologic appearance, and 15 patients (26%) had a difference in visual acuity between fellow eyes of two or more lines on the Snellen distance chart. The asymmetry of visual acuity was present not only in those patients presenting with central lesions but also in those with paracentral and diffuse flecks without a typical central lesion. As the patients were followed over 3 or more years, visual acuities between fellow eyes became more symmetrical. Typically, visual acuity gradually decreases, and once 20/200 is reached, further change rarely occurs (5,48). Almost 50 years after Stargardt’s 1909 report, two of the original patients were reexamined and reported to be essentially unchanged except for an increased area of macular degeneration. Apparently, neither had significant loss of peripheral field nor complaints of night blindness (49). Of course, there are rare patients that do progress to more advanced stages of the disease. Patients that present with the fundus flavimaculatus phenotype are more likely to suffer from severe visual field loss and reduced visual acuity to counting fingers. These patients can eventually have clinical findings that resemble those seen in RP, and they can experience profound functional decline. As further studies are done on genetically identified cohorts, there will be a better understanding of the natural history of this disease.
Important to family members of a STGD1/FF patient, extensive psychophysical testing does not appear to have predictive value as to which family members will develop progressive retinal degeneration. Yet, affected individuals presenting with paracentral and diffuse flecks with or without a typical central lesion (characteristics of FF) have a greater tendency to progress to more advanced stages (5). Armstrong et al. (48) have also supported the concept that characteristics of presentation may be predictive of disease. They concluded that morphologic changes and deterioration of retinal function are more severe in patients with FF than in patients with STGD1 and that the duration of the disease has a greater effect on patients with FF than on patients with STGD1 progression.
It has been shown that a wide variation in clinical phenotype can occur in patients with sequence changes in the ABCA4 gene (7). More recently, 16 patients from 13 families with signs of STGD1/FF and known mutations on both alleles of the ABCA4 gene (15 compound heterozygous, 1 homozygous) were characterized. In the majority of patients, the type and combination of ABCA4 mutations in compound heterozygotes were compatible with the severity of the phenotype as to age of onset and the functional consequences. Unexplained phenotypic differences indicate the influence of other factors (8). The identification of correlations between specific mutations in the ABCA4 gene and clinical phenotypes will facilitate the counseling of patients regarding their visual prognoses. This information will also influence future therapeutic trials for STGD1/FF patients.
Fluorescein Angiography
FA, in the very early stages of STGD1/FF with a beaten-bronze macula, shows a central ovoid area of hypofluorescence surrounded by a zone of blotchy hyperfluorescence. With increasing age, the central lesion may lose the metallic sheen and develop areas of atrophy and hyperfluorescence. The surrounding ring of paramacular white flecks often shows hypofluorescence initially but later hyperfluorescence, indicating staining by the fluorescein dye. The paramacular flecks later disappear leaving hyperfluorescent spots of RPE atrophy. Leakage on fluorescein does not occur unless it is associated with a secondary process, such as choroidal neovascularization.
The peripheral flecks are transient, usually becoming manifest in the second decade but then appearing and disappearing in various locations over several decades. The flecks may hypofluoresce or exhibit a pattern of hyperfluorescence in a zone surrounding the actual flecks. The variation in FA characteristics probably is related to the age of the fleck, the amount of pigment within the fleck, and the degree of RPE involvement (Fig. 19.4).
The lack of choroidal fluorescence (dark choroid) in STGD1/FF has been suggested to be related to the presence of short-wavelength–absorbing chromophores in the RPE layer (50). The phenomenon of dark choroid is evident in approximately half of patients with STGD1/FF (Fig. 19.5). The hypofluorescent choroid appears dark throughout most of the angiogram and may highlight the overlying retinal circulation. The dark choroid effect had been thought to be caused by blockage of the choroidal fluorescence by lipofuscin-laden RPE; however, recent indirect measurements of lipofuscin by autofluorescence have little or no correlation with the presence or lack of a dark choroid on FA (51).
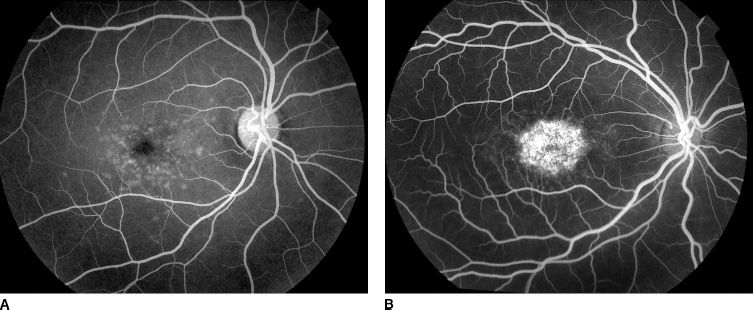
FIGURE 19.5 FA of two different STGD1/FF patients demonstrating the variable phenomenon of the “dark choroid.” A: Shows a uniform choroidal blush that is typical of a “normal” angiogram, whereas (B) shows a relative hypofluorescence of the choroid (dark choroid) that can often highlight the overlying fine retinal capillary structure. Both angiograms are in the late arteriovenous phase. (Courtesy of James Gilman, CRA, Ophthalmic Photography, Department of Ophthalmology, Moran Eye Center, Salt Lake City, UT.)
Aaberg and Han (52) have reported that the presence or absence of choroidal hypofluorescence does not correlate with the phenotypic expression of STGD1/FF. Although there may be considerable variability in phenotypic expression of the disease within a pedigree, there was a more uniform intrafamilial consistency of the dark choroid phenomenon. Moreover, as an individual with STGD1/FF ages, there may be consistent variation in the dark choroid, likely secondary to progressive RPE atrophy.
Other dye-based imaging techniques, such as indocyanine green angiography, may further our understanding of STGD1/FF by revealing different tissue characteristics specifically related to terminal choroidal arterioles and choriocapillaris perfusion defects in the diseased macula (53).
Electrophysiologic Evaluation
Electroretinogram
Abnormalities in the electroretinogram (ERG) parallel the degree of morphologic effect (stage of the disease). The most frequently described ERG abnormality is a decrease in the b-wave amplitude, usually with a normal implicit time. However, in most patients with primarily central macular involvement, the ERG is normal. With increasing amounts of peripheral flecks and central atrophy, the ERG can be moderately reduced. This has been shown to be especially the case in patients with diffuse flecks and vision loss beginning in childhood (54). Comprehensive studies have shown that ERG abnormalities are more prevalent when flecks are clinically evident (55).
Electrooculogram
The electrooculogram is typically normal, but can be reduced when extensive RPE changes are present. The more extensive the disease process, the lower the light peak/dark trough (L/D) ratio (56). Fast oscillations and slow oscillations of the standing potential of the eye are generated by light stimulus–induced hyperpolarization or depolarization of the RPE basal membrane, respectively.
Dark-Adaptation Amplitudes
Dark adaptation can be normal or slightly delayed in STGD1/FF patients, possibly as a result of altered RPE metabolism. Rod amplitudes might need increased time to reach total adaptational amplitudes (57). No frank abnormalities in dark adaptation should be seen in younger, less severely affected patients.
Pattern Visual Evoked Potential
As described earlier in this chapter, rod adaptation kinetics have been reported to be abnormal in patients with STGD1/FF, but cone kinetics have been more recently studied by utilizing pattern visual evoked potential (VEP) recovery testing after macular bleaching. Altered recovery of pattern VEPs after macular bleaching in patients with STGD1/FF was detected in patients with relatively mild clinical impairment (58).
Visual Fields
Visual fields are generally normal, but progressive macular atrophy can give a relative and eventual absolute central scotoma. Annular (ring) scotomata and generalized peripheral field constriction can be seen in patients with more advanced disease and diffuse RPE atrophy.
Color Testing
In a majority of cases, there can be abnormal general color discrimination. Aaberg (5) found an acquired dyschromatopsia of the blue-yellow axis predominant, but noted that all forms of color discrimination dysfunction can be present. Gerth et al. (8) recently evaluated genotypically confirmed STGD1/FF patients and found various color discrimination anomalies in all patients tested. Therefore, color vision dysfunction should not be used as a diagnostic criterion for STGD1/FF, but its presence can help in understanding a patient’s overall visual function. If severely affected color vision is found early in the clinical presentation, the diagnosis of cone dystrophy should be considered.
Optical Coherence Tomography
Optical coherence tomography (OCT) generates highresolution cross-sectional tomograms of ocular tissue, and it has been particularly useful in evaluating and understanding diseases affecting the macula. OCT can be helpful in better understanding the structural changes associated with macular dystrophies. Extensive foveolar thinning and macular volume loss can be observed in STGD1/FF. Ultrahigh-resolution OCT scans of patients with STGD1/FF showed a diffuse retinal atrophy with a reduction in the thickness of all retinal layers, sub-RPE deposits corresponding with the flecks, and a focal loss of photoreceptor inner and outer segments. Loss of the photoreceptor layer on ultrahigh-resolution OCT has been found to correlate with visual acuity and to be a better indicator of visual function than retinal thickness (59). Modern OCT technology also allows the visualization of deposits, very likely lipofuscin, within the RPE cells (60).
Additionally, despite the fact that STGD1/FF is primarily an outer retinal and RPE degeneration, retinal nerve fiber layer (RNFL) thickness has been shown to be reduced in approximately 50% of patients with STGD1/FF. Patients with preserved RNFL may prove to be better candidates for restorative therapies in the future (61).
Fundus Autofluorescence
The lipofuscin or lipofuscin-like material that accumulates in RPE cells has intrinsic autofluorescence properties. One of the main fluorophores within lipofuscin is A2E (32). Spectral characteristics of in vivo FAF (relatively low excitation wavelengths of 510 to 550 nm with emission between 630 and 750 nm) are consistent with those of lipofuscin (62). Confocal scanning laser ophthalmoscopes (SLOs) and fundus cameras with specialized filter sets have been used to detail autofluorescence as an index of lipofuscin in the living eye. The normal fundus shows a consistent pattern in patients older than 6 years, with diffuse autofluorescence that is most intense between 5 and 15 degrees from the fovea center (63).
Heritable macular dystrophies, such as STGD1/FF, have been shown to have more autofluorescence consistent with accelerated lipofuscin accumulation (51). The entire fundus of patients with STGD1/FF shows abnormally intense autofluorescence, suggesting a generalized biochemical abnormality of the RPE despite the discrete flecks seen on clinical examination. Interestingly, when correlating FA with FAF, high levels of autofluorescence may be present with or without the classic finding of a dark choroid on FA (51).
Focal areas of hyperautofluorescence correspond to areas of increased lipofuscin density and poor RPE function and may be predictive of future hypoautofluorescence and RPE atrophy (32). Areas of RPE attenuation or atrophy display a loss of autofluorescence, and this makes autofluorescence imaging valuable in assessing general RPE attrition. However, areas of hypoautofluorescence only correlate with scotomata on macular perimetry approximately 50% of the time, possibly because areas of RPE attenuation still maintain functioning overlying photoreceptors (64). If followed over time, an area of attenuated RPE likely would atrophy and cause a corresponding scotoma. Areas of RPE atrophy and corresponding hypoautofluorescence on FAF have been shown to have overlying loss of photoreceptors by using adaptive optics SLO technology (65).
Interestingly, foveal macular pigment has been related to foveal cone acuity in STGD1/FF, and these pigments may serve as reliable markers for the presence of foveal cones. Infrared light may serve as a sensitive screening tool for detection of early STGD1/FF (66). Near-infrared autofluorescence (NIA) imaging utilizes longer excitation wavelengths of light to highlight autofluorescent properties of other molecular structures within the eye, most likely melanin. NIA imaging may detect the pathologic changes seen in STGD1/FF earlier than standard FAF (32).
Studies utilizing single-wavelength ultraviolet-absorbance high-performance liquid chromatography have examined the autofluorescence properties of A2E in a rat model (67). Calibration of an SLO to this wavelength might allow for in vivo detection of damaging A2E accumulation to assess disease progression.
Genetic Analysis
Allikmets et al. (16) designed a genotyping microarray (gene chip) for the ABCA4 gene to enable comprehensive screening of populations with ABCA4-associated retinal pathology, including STGD1/FF. The chip includes more than 400 of the sequence variants that have been described in this gene, allowing the detection of all these variants in one reaction. The ABCR400 array was constructed by the allele-specific primer extension technology (described at http://www. asperbio.com). In tested STGD1/FF patient cohorts, the array detected between 54% and 78% of all possible disease-associated alleles (10). Commercially available Stargardt genetic testing now utilizes both microarray testing and sequence analysis of the entire coding region of the ABCA4 gene, and more than 550 mutations have been identified.
Differential Diagnosis
In more advanced stages of STGD1/FF that usually present in adult life, the differential diagnosis is extensive. For a comprehensive review of macular dystrophies, readers can refer to Dr. Gass’ Stereoscopic Atlas of Macular Disease (68). Genetic characterization is aiding in routine diagnosis of STGD1/FF, but until these techniques are readily available and shown to be reproducible and reliable, clinical findings remain the mainstay for making the diagnosis. For the purposes of this pediatric retina reference, the clinical differential can be limited to other macular diseases that typically present in childhood, including but not limited to dominant Stargardt-like macular dystrophy [STGD2 13q34 (69), STGD3 6q11-q15 (70), Kniazeva (71), STGD4 4p (72)], cone–rod dystrophy, pericentral RP, pattern dystrophy, vitelliform dystrophy (Best disease), X-linked juvenile retinoschisis, dominant cystoid macular dystrophy, solar retinopathy, acute macular neuroretinopathy, central areolar choroidal dystrophy, and fundus albipunctatus.
Trauma
RPE cells engorged with lipofuscin may predispose patients with STGD1/FF to subretinal fibrosis and outer retinal damage in the setting of minor contusion trauma (73). Sports activities should minimize contact-related activities when possible. The incidence of minor blunt trauma may be reduced with polycarbonate safety glasses.
Motor vehicles present their own problems. Szlyk et al. (74) have shown that the proportion of individuals with juvenile macular dystrophies, including STGD1/FF, involved in accidents was comparable to matched controls. However, for 13 of the 20 subjects with central vision loss who did not restrict their driving to daylight hours, there was a greater likelihood of involvement in nighttime accidents than in the control group. Motor vehicle accidents can be complicated by secondary trauma associated with airbag safety devices (75). Airbags combined with seat belts are an effective means of reducing injury and death during motor vehicle accidents. It is hoped that safetyindustry researchers will be able to develop modifications that can continue to save lives and minimize ocular risk.
Medical/Surgical
In addition to limitations of central vision inherent to the natural course of STGD1/FF, patients may also suffer from secondary processes that further limit visual potential. Choroidal neovascularization has been reported in patients with STGD1/FF (76). Photodynamic therapy has been employed to retard progression of subfoveal choroidal neovascular membrane in a patient with STGD1/FF (77). Potter et al. (78) have reported surgical intervention for a patient with STGD1/FF who suffered further visual impairment from a prominent epiretinal membrane. The authors caution that surgical intervention in eyes with preexisting degenerative changes may lead to atypical complications or outcomes.
School-Age Issues
In children and teens with STGD1/FF, it is very important to employ adequate low-vision care and modify behavioral and environmental factors to maximize visual performance. For example, these students may have light and glare control problems in their classrooms. Adaptations such as sitting away from the window or shutting the curtains may be necessary. Moreover, these students should have permission from school administrators to wear special sun filters (sunglasses) in classrooms and on campus when needed to decrease glare and light sensitivity.
Because the vision loss may progress over time, each student should have a teacher of the visually impaired (VI). A VI teacher can assess the classroom and educational plan for the child. With the low-vision specialist’s recommendations, the VI teacher makes adaptations including low-vision devices, large-print materials, and other special services the child will need each year in school. Some students may require extended time on reading assignments and tests. Computer software is also available to convert standard programs into more user-friendly formats for people with low vision.
As mentioned earlier, there are also safety issues to address in young active students. Eyewear with polycarbonate lenses should be worn for protection of the eyes from unexpected injury. Counseling includes safety issues in physical education. Low-impact and minimal contact sports such as swimming and track are good options. Sports with fast-moving projectiles may put young patients at undo risk. Protective face shields are essential if patients are to participate in such sports.
Preventive steps should be exercised when this condition manifests during early life. However, it should be noted that STGD1/FF rarely causes legal blindness during childhood years.
Low-Vision Care
Young patients with STGD1/FF respond well to magnification, and simple bifocals may be used in the early stages. In later stages, closed-circuit television systems are helpful. Mobility is usually minimally affected. Some young adults with STGD1/FF can drive with the assistance of bioptic devices, but it may be for a limited time. In deciding on low-vision devices for children and especially adolescents, it is important to maintain good cosmetic appearance.
ROLES OF OTHER PHYSICIANS AND HEALTH CARE PROVIDERS
Since the Americans with Disabilities Act passed in 1990, attempts have been made to determine the functional limitations imposed by various types of visual impairment. Szlyk et al. (79) have better defined real and perceived functional limitations of patients with juvenile macular degeneration. Their work has provided insight into the difficulties of performing everyday tasks in these patients. From a clinical perspective, it is important to be aware of the varying profiles of functional impairment produced by different visual disorders in order to be more sensitive to the individual needs of patients with vision loss. It is also important for the clinician to be prepared to direct patients and parents to information and support resources at the time of diagnosis to minimize uncertainty and emotional distress (Table 19.1).
TABLE 19.1
Information and support sites for STGD1/FF patients on the World Wide Web
STGD1/FF, Stargardt disease and fundus flavimaculatus.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


