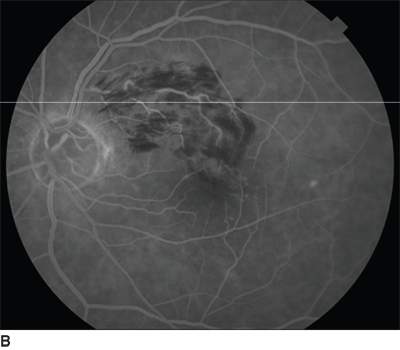
FIGURE 18.1. Fundus photo (A) and FA (B) of acute presentation of BRVO showing dilated and tortuous vein surrounded by wedge-shaped area of deep and superficial retinal hemorrhages and retinal edema. The areas of hyperfluorescence superior to the foveal center are due to ME.
The visually significant complications include ME (most common), nonperfusion, and vitreous hemorrhage from neovascularization.5,8,9 In patients with perfused ME, approximately one third will regain some vision spontaneously. However, this becomes less likely the longer the edema persists. Early in the course of BRVO, macular nonperfusion can result in permanent vision loss.8 Large areas of persistent retinal nonperfusion can lead to neovascularization of the retina or disc and can cause vitreous hemorrhage. In the Branch Vein Occlusion Study (BVOS), retinal neovascularization developed in 36% of eyes with five disc diameters or more of retinal nonperfusion.11 It is important to recognize that perfused BRVO may progress to the nonperfused type during the ensuing weeks, months, and even years.
Diagnostic Testing
Intravenous fluorescein angiography (FA) and optical coherence tomography (OCT) can assist in the evaluation of BRVO. Angiographic findings reflect changes in vessel permeability, caliber, and patency and assist in identifying areas of ME, neovascularization, and nonperfusion. OCT is a useful tool for the detection and monitoring of ME in BRVO. The findings from the Standard Care versus COrticosteroid for REtinal Vein Occlusion (SCORE) Study revealed a modest correlation between OCT-measured center point thickness and VA.12
Treatment
As our understanding of the disease improves, the standard of care is likely to continue to evolve. Available data concerning many of the reported treatment modalities are based on case series without controls or randomization. Initial evaluation may include testing for hypertension, fasting blood glucose level, complete blood count with differential and platelets, coagulation studies, and erythrocyte sedimentation rate. Follow-up generally consists of ophthalmologic evaluation every 1 to 3 months for the first year, followed by every 3 to 12 months thereafter. Grid laser photocoagulation for ME, (BRVO), and scatter laser photocoagulation for retinal or anterior segment neovascularization (BRVO and CRVO) continue to remain the mainstay of treatment.10,13 Pharmacotherapy is currently used by some clinicians as an adjunct or replacement for laser photocoagulation until data are available from ongoing clinical trials on the use of intravitreal drugs as primary therapy (Figure 18.2).
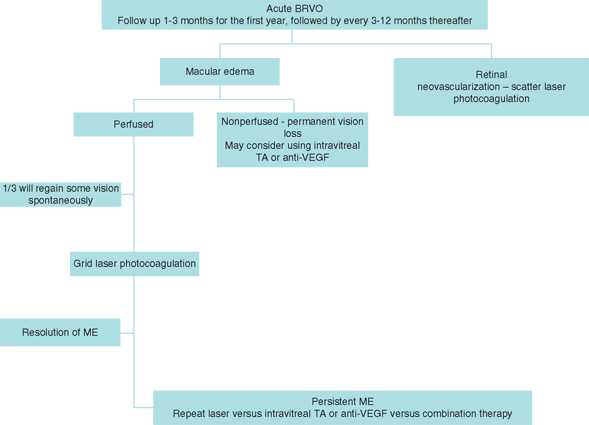
FIGURE 18.2. Stepwise approach for pharmacotherapy of BRVO.
Intraocular Corticosteroids
Intravitreal triamcinolone acetonide (TA) has been investigated for persistent ME in RVO and has been shown experimentally to reduce the breakdown of the blood-retina barrier by inhibiting factors such as prostaglandins, interleukins, VEGF, and protein kinase C. Small case studies have shown favorable anatomical response to intravitreal injection of TA within several weeks, as demonstrated by OCT images.14–18 However, a favorable VA response may be more likely in patients with perfused rather than nonperfused ME. Retreatment may also be performed in some patients due to recurrent ME. A human pharmacokinetics study of nonvitrectomized eyes found a single 4-mg intravitreal injection of triamcinolone to have a mean half-life of 18.6 days with measurable concentrations expected to last approximately 3 months.19 Reported side effects include cataract, increased intraocular pressure (IOP), and injection-related complications including noninfectious and infectious endophthalmitis, retinal detachment, vitreous hemorrhage, and lens injury.14–18
The SCORE (Standard care vs. COrticosteroid for REtinal Vein Occlusion) study, a multicenter, randomized, Phase 3 National Eye Institute–sponsored study, elucidated the efficacy and safety of standard care versus intravitreal injection(s) of TA for ME secondary to BRVO and CRVO. Individuals with BRVO or CRVO with associated ME of up to 24 months’ duration and best-corrected VA between 19 and 73 ETDRS letters (corresponding to approximately 20/40 to 20/400 Snellen VA) were eligible for participation in the SCORE study.20 The two primary study objectives of the SCORE-BRVO trial were (a) to determine whether intravitreal TA at 1 and 4-mg doses produces greater visual benefit, with an acceptable safety profile, than grid photocoagulation (standard care), when appropriate, for the treatment of vision loss associated with ME secondary to BRVO and (b) to compare the efficacy and safety of the 1 and 4-mg TA doses. The results of the SCORE-BRVO trial21 demonstrated no significant differences among the three treatment groups for a gain in VA letter score of 15 or more at 12 months (29%, 26%, and 27% in the standard care, and 1 and 4-mg TA groups, respectively). An early positive treatment response of a gain in VA letter score of 15 or more was observed at month 4 in the 4-mg TA group compared with the 1-mg TA and standard care groups. After month 12 and through month 36, the mean improvement from baseline VA letter score was greatest in the standard care group compared with the two TA groups. With respect to OCT-measured center point thickness, all three groups showed a decrease from baseline to month 12. Analogous to the VA results, only at month 4 did the 4-mg TA group demonstrate a greater treatment effect on center point thickness than the 1-mg and standard care groups; at all other times investigated (months 8–36), the standard care group demonstrated the greatest overall median decrease in center point thickness from baseline. The rates of adverse events were higher in the 4-mg TA group compared with the 1-mg and standard care groups. There was a dose-dependent higher frequency of initiating IOP-lowering medications in the TA groups (41% in 4 mg, and 8% in 1 mg) compared with the standard care group (2%). The proportion of phakic eyes that had new-onset lens opacity or progression of an existing opacity through 12 months based on assessment at the clinical center was greater in the two TA groups (35% in 4 mg, and 25% in 1 mg) compared with the standard care group (13%). Most cataract surgeries were performed during the second year of the study and occurred with the highest frequency in the 4-mg group (n = 35). The rates of adverse events with respect to cataract surgery and elevated IOP were similar between the standard care and 1-mg groups. Thus, the SCORE study results support grid photocoagulation as the continued standard of care for patients with decreased VA associated with ME secondary to BRVO up to 12 months and possibly up to 36 months.21 Furthermore, the results of this trial also substantiate that grid photocoagulation must be the benchmark against which other treatments for vision loss associated with ME secondary to BRVO should be compared in clinical trials (Box 18.1).
BOX 18.1 SCORE Trial in BRVO
- Purpose: This multicenter, randomized clinical trial compared the efficacy and safety of 1- and 4-mg doses of preservative-free intravitreal triamcinolone with standard care (grid photocoagulation in eyes without dense macular hemorrhage and deferral of photocoagulation until hemorrhage clears in eyes with dense macular hemorrhage) for eyes with vision loss associated with ME secondary to BRVO.
- Conclusions: There was no difference identified in VA at 12 months for the standard care group compared with the triamcinolone groups. Rates of adverse events (particularly elevated intraocular pressure and cataract) were highest in the 4-mg group. Grid photocoagulation as applied in the SCORE Study remains the standard care for patients with vision loss associated with ME secondary to BRVO. (SCORE Study Group, Arch Ophthalmol. 2009;127:1115).
Ozurdex is an extended delivery bioerodable dexamethasone PLGA (polylactic acid polyglycolic acid) copolymer complex, which has been also studied in persistent ME secondary to RVO.22 The release of dexamethasone depends on the diffusion through the polymer matrix, solubility of the polymer, and rate of degradation of polymer matrix. The drug is released for approximately 35 days, and the polymer matrix is absorbed in 60 to 180 days. A specially designed proprietary instrument is used for the intravitreal injection of the Ozurdex implant, thus obviating the need of surgery.23 It consist of 22-g hypodermic needle, 1 inch long mounted on a device measuring 165 × 13 mm preloaded with the implantable drug product. The drug is injected at the inferior pars plana by pressing a button. Common adverse events associated with implant procedure include anterior chamber flare, conjunctival hyperemia, conjunctival hemorrhage, eye pain, floaters, IOP increase, vitreous hemorrhage, and vitritis.23 The efficacy of the Ozurdex implant was tested in a 6-month randomized phase 2 trial for treatment of persistent ME (>90 days) due to various causes including RVO.22 The VA at entry was 20/40 to 20/200, and the patients were randomly assigned to one of the three treatment regimens: a single Ozurdex implant containing 350 µg of drug, 700 µg of drug or observation. There was statistically significant improvement in best-corrected VA of 10 letters or more in 35% of patients in the 700-µg dose (p < 0.001) at day 90 and the drug was well tolerated for 180 days. There was no cataract progression, and a transient IOP elevation was observed at the different time points, which were readily managed with topical antiglaucoma medications. The dose of 700 µg seemed to be effective and may have potential as new therapy for recalcitrant ME secondary to RVO. A phase 3 clinical trial evaluating the Ozurdex implant for RVO has been completed, and study results are currently pending (Box 18.2).
BOX 18.2 OZURDEX in Retinal Vein Occlusions
Ozurdex (dexamethasone intravitreal implant), 0.7 mg (Allergan), has been approved by the U.S. FDA as the first drug therapy for ME in BRVO or CRVO. It is administered via intravitreal injection via an innovative NOVADUR solid polymer delivery system. The dexamethasone drug delivery system (DDS) is composed of a biodegradable copolymer of lactic acid and glycolic acid. The polymer slowly biodegrades into carbon dioxide and water, as dexamethasone is released. Since the implant eventually dissolves completely, there is no need for surgical removal. Two multicenter, double-masked, randomized clinical trials showed that the time to achieve a ³15 letter (3-line) improvement in best-corrected visual acuity (BCVA) cumulative response rate curves was significantly faster with the Ozurdex implant compared to sham (p < 0.01) with Ozurdex-treated patients achieving a 3-line improvement in BCVA earlier than sham-treated patients. The onset of effect with Ozurdex, defined as a ³15 letter (3-line) improvement in BCVA, occurred within the first 2 months after implantation in approximately 20% to 30% of subjects. The duration of effect persisted approximately 1 to 3 months after onset. The most common ocular adverse reactions reported by >2% of the patients in the first 6 months included increased intraocular pressure (25%), conjunctival hemorrhage (20%), eye pain (7%), conjunctival hyperemia (7%), ocular hypertension (4%), cataract (4%), and vitreous detachment (3%), and headache (3%).
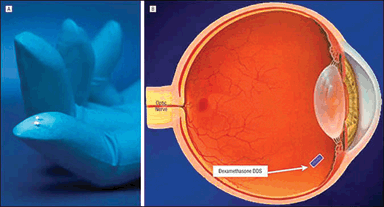
Dexamethasone drug delivery system (DDS). A: Dexamethasone DDS implants. The larger implant is the 700-µg dose, and the smaller implant is the 350-µg dose. B: Approximate location of the dexamethasone DDS (indicated with the arrow; shown larger than actual size for illustrative purposes) after insertion into the eye. (Reproduced from Kuppermann BD, Blumenkranz MS, Haller JA, et al. Randomized controlled study of an intravitreous dexamethasone drug delivery system in patients with persistent macular edema. Arch Ophthalmol. 2007;125(3):309–317, with permission.)
Periocular Corticosteroids
Periocular application of TA has been also investigated for treatment of ME associated with BRVO. In a randomized study, 60 patients received either a single intravitreal injection (4 mg) or repeated retrobulbar injections (40 mg, three times) of TA, first injection given approximately 1 week after focal laser photocoagulation.24 The foveal thickness and VA improvements were significantly better after intravitreal injection, and the need for reinjections was significantly greater in the retrobulbar group.
Anti-Vascular Endothelial Growth Factor Antibodies
Intravitreal VEGF levels have been shown to be increased in patients with ME with BRVO and are correlated significantly with the area of nonperfusion and the severity of ME. A number of case reports, small retrospective or prospective noncontrolled studies, describe improved retinal thickness and VA following intravitreal bevacizumab.25–29 In one series, the best-corrected VA (BCVA) improved from a baseline mean of 20/333 to 20/91 at 4 weeks after one injection of bevacizumab. However, the effect was not sustained, and at 12 weeks, the VA was reduced to 20/126.30 A recent prospective clinical trial showed an increase in mean VA from 50 letters (20/100) at baseline to 66 letters (20/50+1; +16 letters; p < 0.001) at month 12 and central retinal thickness (CRT) decreased from 558 µm at baseline to 309 µm at month 12 (-249 µm; p < 0.001) in 21 eyes with persistent ME (>3 months) treated with three initial intravitreal bevacizumab injections of 1 mg at a monthly interval.31 In this study, retreatment was based on CRT, and if continuous injections were needed up to month 6, the dose was increased to 2.5 mg. However, the ME had not resolved completely in 86.2% of all patients after three initial injections. Furthermore, the retreatment rate was high with a mean of 8 out of 13 possible injections over 12 months. There was recurrence of ME after initial improvement in many patients, and 20.7% of all patients showed no complete resorption of ME despite continuous treatment for up to 1 year and an increase of the dose to 2.5 mg after month 6. A 12-month randomized double-masked, sham injection–controlled Phase 3 study (BRAVO) of 397 patients designed to assess the safety and efficacy profile of ranibizumab (Lucentis) in ME secondary to BRVO is currently underway. It consists of a 6-month, sham-controlled treatment period, followed by a 6-month observation period (during which all participants are eligible to receive Lucentis as needed). During the first 6-month period, participants received monthly injections of either 0.3 or 0.5 mg of Lucentis or monthly sham injections. All participants who met prespecified criteria were eligible to receive rescue laser treatment during the first 6-month period. Results at 6 months showed 55.2 percent of patients who received 0.3 mg of Lucentis, and 61.1% who received 0.5 mg of Lucentis had their vision improved by 15 letters or more on the study eye chart compared to 28.8% of patients receiving sham injections (data obtained from Genentech press release on October 4, 2009). Mean gain in BCVA was observed beginning at day 7 with a 7.6- and 7.4-letter gain in the 0.3 and 0.5 mg study arms of Lucentis, respectively, compared with 1.9 letters in the sham injection arm (Box 18.3).
BOX 18.3 Anti-VEGF Trials in BRVO and CRVO
The US FDA has approved ranibizumab for treatment of macular edema to retinal vein occlusions based on data from the BRAVO and CRUISE trials.
BRAVO Trial
- Purpose: A 12-month randomized double-masked, sham injection-controlled Phase 3 study designed to assess the safety and efficacy profile of ranibizumab in ME secondary to BRVO is ongoing. During the first 6-month period, participants received monthly injections of either 0.3 or 0.5 mg of ranibizumab or monthly sham injections.
- Results: Results at 6 months showed 55.2% of patients who received 0.3 mg of ranibizumab and 61.1% who received 0.5 mg of ranibizumab had their vision improved by 15 letters or more on the study eye chart compared to 28.8% of patients receiving sham injections. (Data obtained from Genentech press release on October 4, 2009.)
CRUISE Trial
- Purpose: A randomized clinical trial to substantiate the effect and safety of ranibizumab in patients with ME due to CRVO is ongoing. Patients received monthly injections of either 0.3 or 0.5 mg of ranibizumab or monthly sham injections for a 6-month period, followed by a 6-month observation period (during which all participants are eligible to receive ranibizumab as needed).
- Results: At 6 months, 46.2% of patients given 0.3 mg of ranibizumab and 47.7% given 0.5 mg of ranibizumab had their vision improved by 15 letters or more compared to 16.9% of patients receiving sham injections. (Data obtained from Genentech press release on October 4, 2009.)
Thus, the role of antivascular endothelial growth factor (anti-VEGF) agents in the treatment of BRVO has yet to be determined. Long-term results and the number of treatments required are unknown. A definitive comparative study of focal laser versus anti-VEGF agents is yet to be performed. Anti-VEGF agents may be a reasonable treatment option when the ME is refractory to laser treatment (Box 18.4) and (Fig. 18.3).
BOX 18.4 Other Clinical Trials in Retinal Vein Occlusions
COPERNICUS Trial: Vascular Endothelial Growth Factor (VEGF) Trap Eye: Investigation of Efficacy and Safety in CRVO
- Purpose: A randomized, double-masked, controlled Phase 3 study of the efficacy of VEGF Trap-Eye (Regeneron) injected into the eye on vision function in subjects with ME as a consequence of CRVO. Arm 1: Monthly IVT injection of VEGF Trap-Eye 2.0 mg until Week 24 Primary Endpoint. Arm 2: Monthly Sham IVT injection until week 24 Primary Endpoint.
- Status: Recruitment phase.
GALILEO Trial: Vascular Endothelial Growth Factor (VEGF) Trap-Eye: Investigation of Efficacy and Safety in CRVO
- Purpose: A randomized, double-masked, sham-controlled Phase 3 Study of the efficacy of repeated intravitreal administration of VEGF Trap-Eye in subjects with ME due to CRVO. Arm 1: Intravitreal injection. Weeks 0 to 20 injection of VEGF Trap-Eye every 4 weeks; weeks 24 to 48 every 4 weeks reassessment and either (PRN) injection of VEGF Trap-Eye or sham injection; weeks 52 to 100 safety follow-up. Arm 2 Control : Sham treatment. Weeks 0 to 20 sham treatment every 4 weeks; weeks 24 to 48 every 4 weeks reassessment and sham injection; weeks 52 to 100 safety follow-up.
- Status: Recruitment phase.
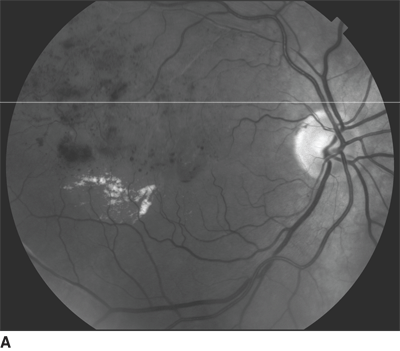
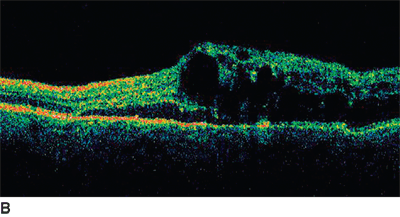
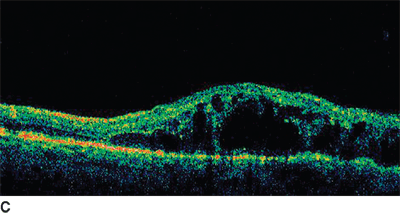
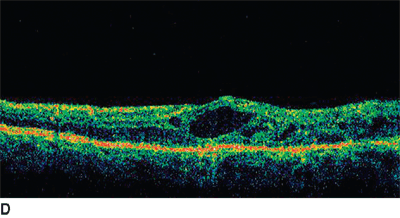
FIGURE 18.3. Fundus photo (A) and baseline OCT (B) of patient with chronic BRVO and ME. The patient underwent laser photocoagulation; however, there was no resolution of ME as noted in follow-up OCT (C). Hence, the patient received intravitreal injection of bevacizumab at 8 months after laser and a second injection 6 weeks after the first injection. There was a considerable resolution of ME as noted on OCT (D) following the two injections.
Recombinant Tissue Plasminogen Activator
The pathophysiologic basis for using recombinant tissue plasminogen activator (r-tPA) in BRVO is related to its thrombolytic effects. The additional effects caused by r-tPA include induction of posterior vitreous detachment (PVD) and development of collateral vessels. In a retrospective interventional case series, seventeen eyes presenting with ME caused by BRVO were treated with an intravitreal r-tPA injection.32 The mean logMAR VA significantly improved from 0.603 ± 0.327 at baseline to 0.388 ± 0.248 (p < 0.01) at 1 month and 0.359 ± 0.319 (p < 0.05) at 6 months. The mean foveal thickness significantly decreased from 738 ± 156 µm at baseline to 454 ± 213 µm (p < 0.001) at 1 month and 253 ± 164 µm (p < 0.001) 6 months. The complications noted in this study were a focal pigmentary alteration sparing the macula in one case and a retinal hole after PVD induced by r-tPA in another case. The efficacy of r-tPA in treatment of BRVO is still unclear, given the absence of randomized trial comparing it with grid laser photocoagulation.
Systemic Therapies
Systemic anticoagulation
Systemic anticoagulation including oral acetylsalicylic acid, subcutaneous heparin, or intravenous thrombolysis has not been shown to be of ocular benefit in patients with BRVO and may potentially worsen concurrent or future intraretinal hemorrhage. Troxerutin has been studied in double-blind randomized study of 26 patients with BRVO <5 months.33 This drug acts by inhibiting erythrocyte and platelet aggregation and improves erythrocyte deformability, and thereby retinal microcirculation. In this study, baseline visual acuities were well matched, and patients receiving Troxerutin treatment had a mean VA of 20/40 or better than the placebo group, although this difference was not found to be statistically significant. This study was limited by a small sample size, a short randomized, controlled, and masked follow-up period (4 months). Ticlopidine is an inhibitor of platelet aggregation and was compared with placebo in treating patients within 3 weeks from the onset of symptoms for BRVO. At 6-month follow-up, there was a significant difference between the Ticlopidine treatment and placebo groups for VA.33 However, the Ticlopidine treatment group was associated with gastrointestinal symptoms and skin reactions.
Isovolemic Hemodilution
BRVO has been reported to be associated with hyperviscosity due to higher hematocrit and plasma viscosity. Studies have demonstrated that hypervolemic or isovolemic hemodilution, commenced within 3 months of the onset of symptoms of a BRVO, accelerates the rate of visual recovery and also has a positive effect on the final VA at 1 year.34 However, reported complications of hemodilution include deep vein thrombosis and hypotension.
CENTRAL RETINAL VEIN OCCLUSION
Pathophysiology
The pathogenesis and treatment of CRVO continue to be highly debated. Hayreh has suggested that perfused CRVO occurs after occlusion of retinal venous flow, while nonperfused CRVO develops after occlusion of venous and arterial flow at their entry into the optic nerve based on animal models.35 A histopathologic study noted fresh or a recanalized thrombus in enucleated eyes with chronic, nonperfused CRVO, and neovascular glaucoma.36 Furthermore, vasculitis, thrombophilic factors, and hyperviscous states have been associated with CRVO.37 An increased risk of CRVO has been noted in eyes with open-angle glaucoma. Hypoxia-induced upregulation of VEGF appears to play an important role in the development of neovascularization and ME secondary to CRVO.38 Inhibition of VEGF in a nonhuman primate model of CRVO can prevent the development of iris and angle neovascularization.39 VEGF induces expression of thromboplastin, a potent procoagulant tissue factor that may aggravate the retinal ischemia induced by the CRVO.40 Furthermore, VEGF induces retinal endothelial swelling, which may lead to further capillary nonperfusion.41
Clinical Features
Classically, a CRVO presents with marked dilation and tortuosity of the retinal veins, extensive retinal edema, pronounced superficial and deep retinal hemorrhages radiating outward from the optic disc in all quadrants, cotton-wool spots, and optic disc swelling. Typically, two thirds of all CRVOs are perfused, while the remaining one third are nonperfused.42 Nonperfused CRVO is often more dramatic in its presentation and is often associated with a relative afferent pupillary defect, and VA worse than 20/400. Overlying retinal hemorrhage may impede the ability to evaluate the underlying capillary perfusion status. One third of perfused CRVOs progress to nonperfused over the course of the disease. Risk factors for progression include worse presenting VA, severe ME, and progressive intraretinal hemorrhage.
The clinical course after a CRVO is highly variable.42 The retinal hemorrhages may resolve over weeks or may persist for years. The venous dilatation and tortuosity typically resolve with time, and marked fibrous sheathing of the retinal veins and arteries may develop. The disc swelling slowly resolves and may develop disc pallor in nonperfused cases. Optociliary shunt vessels develop commonly following CRVO and may be associated with a decreased risk of developing anterior segment neovascularization. Microaneurysms are a frequent finding following CRVO, whereas macroaneurysms and hard exudates are less common. ME is common following CRVO and may resolve quickly, persist chronically, vary intermittently, or develop late in the course of the disease. Neovascularization typically occurs on the iris in CRVO; however, neovascularization may develop on the disc and/or retina. If neovascularization develops, it usually is present within the first 7 months of the CRVO. The rate of iris neovascularization is 10% in perfused eyes and is 45% to 80% in nonperfused eyes. Of eyes that develop iris neovascularization, approximately two thirds will develop neovascular glaucoma without treatment.13,43
Diagnostic Testing
Clinical examination combined with FA is most commonly used to determine perfusion characteristics. The CVOS used 10 disc areas of nonperfusion as its cutoff for predicting risk.13 In ME, FA shows parafoveal and paramacular capillary leakage that appears as hyperfluorescence that increases in size and intensity with time, often in a petalloid type pattern, and is a risk factor for progression of perfused CRVO to nonperfused CRVO. Nonperfusion near the fovea is associated with worse visual prognosis. Electrophysiologic testing may be helpful in the evaluation process when FA cannot demonstrate the extent of nonperfusion (e.g., in cases with extensive intraretinal hemorrhage). ERG b/a-wave amplitude ratios, photopic and scotopic b-wave amplitudes, and flicker amplitudes were significantly smaller in eyes with extensive capillary nonperfusion than in perfused eyes.44 The OCT can aid the clinician in evaluating ME, epiretinal membrane formation, and subretinal fluid accumulation following CRVO.
Treatment
While early diagnosis of a CRVO may not require much effort on the part of the clinician, predicting the clinical course and choosing treatment options can be much more difficult and challenging (Figure 18.4). A complete ocular examination, including IOP measurement, slit-lamp biomicroscopy, gonioscopy to rule out neovascularization of the iris or angle, and a dilated fundus examination, is recommended at the initial presentation and at monthly follow-up examinations during the first 9 months. The Central Vein Occlusion Study (CVOS) was a landmark study that recommended prompt application of panretinal photocoagulation after the development of iris or angle neovascularization in eyes with nonperfused CRVO. However, the study showed no benefit from grid laser photocoagulation for CRVO-associated ME.13 Laser-induced chorioretinal venous anastomosis and radial optic neurotomy have also been shown to improve visual status, but they may be associated with potentially serious complications and have not been evaluated in controlled randomized trials. Various pharmacologic agents have been used as solo or as combination therapies primarily for management of macula edema.
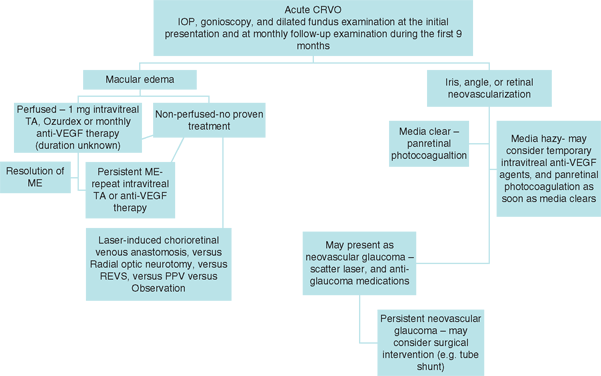
FIGURE 18.4. Stepwise approach for pharmacotherapy of CRVO.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


