cGMP, cyclic guanosine monophosphate; RPGR, retinitis pigmentosa GTPase regulator; PDE, phosphodiesterase enzyme; ATP, adenosine triphosphate.
Presenting symptoms of cone–rod dystrophy include decreased central vision, dyschromatopsia, and light sensitivity (10). Patients typically report that they are most comfortable in low ambient light settings such as dawn or twilight. Early in the course, patients may not have nyctalopia, but with progression of disease, they will develop nyctalopia and peripheral vision loss. Typically, fundus changes are similar to RP with bone spicules and retinal pigment epithelium (RPE) atrophy. However, fundus changes tend to occur in the macula and posterior pole before the retinal periphery. Macular changes include geographic atrophy, a beaten bronze appearance, a bull’s-eye maculopathy, and a tapetal sheen (Fig. 18.1).
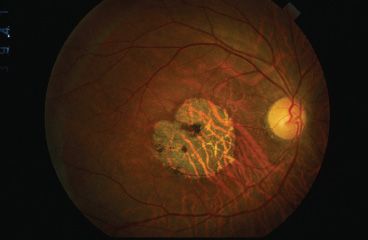
FIGURE 18.1 Cone–rod dystrophy. Patient with an X-linked cone–rod dystrophy. There is a subtle tapetal sheen temporally, and the patient has a geographic area of central macular atrophy.
The characteristic ERG demonstrates greater involvement of the cone system than the rod system with full-field testing. One form of recessive cone–rod dystrophy caused by mutations in the KCNV2 gene is characterized by the typical reduction in cone function and a concomitant supranormal rod response (11). Further attempts have been made to characterize cone–rod dystrophies based on their dark-adapted perimetry characteristics (12). Finally, the multifocal ERG is diffusely abnormal and may be nonrecordable in patients with cone–rod dystrophies. Patients with focal macular changes, such as seen in Stargardt disease, will have greater central loss (Fig. 18.2).
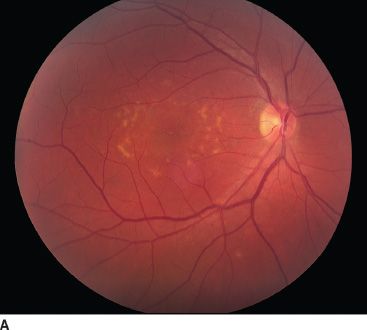
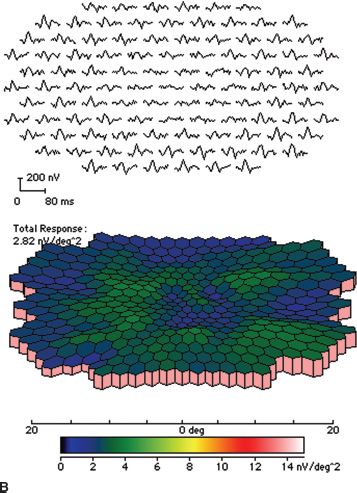
FIGURE 18.2 Stargardt disease. A: Note the classic appearance of pisciform flecks in the macula and beaten bronze appearance centrally. B: Multifocal ERG. The top image represents trace arrays for the central 40 degrees of the patient’s field of vision. Note the loss of amplitude centrally. This is depicted in a graphic format in the bottom image as a response density plot. The central depression represents the loss of function due to Stargardt disease. The peripheral, shallower depression represents the blind spot.
Clinical diseases once believed to be separate from cone–rod dystrophies are now included within the broad group because of shared electrophysiologic characteristics. Foremost among these is Stargardt disease. The presence of cone dysfunction has been noted to be mild to severe and may present at any point in the course of a classic clinical appearance of Stargardt disease/fundus flavimaculatus. Although derangements in ERG function are more common in late stages of this disease, patients have presented with minimal fundus changes and severe cone dysfunction early in the course of the disease. Lois et al. (7) suggested that the presence of advanced ERG derangements might be an indicator of a poor long-term outcome. Itabashi et al. (13) reported that patients with a clinical phenotype of fundus flavimaculatus, that is, pisciform changes involving the midperipheral and posterior retina rather than disease limited to the macula, tended to present with worse ERG dysfunction. Stargardt disease is dealt with in detail elsewhere in this textbook (Chapter 19).
Another condition with severe cone dysfunction at presentation is fundus albipunctatus. Although fundus albipunctatus is classically considered to be nonprogressive, some patients will also develop cone dysfunction accompanied by macular changes in midadulthood (14) (see Congenital Stationary Night Blindness).
Systemic disease associations with cone–rod dystrophies are rare. Spinocerebellar ataxia has been associated with cone–rod dystrophies (15). There are two reported cases of thiamine-responsive megaloblastic anemia and cone–rod dystrophy (16) and a reported case of achondroplasia with cone–rod dystrophy (17). Alström syndrome also demonstrates a cone–rod dystrophy phenotype with cardiomyopathy and type 2 diabetes mellitus (18). The CNNM4 gene is associated with Jalili syndrome, which includes autosomal recessive cone–rod dystrophy, amelogenesis imperfecta, and neurofibromatosis type 1 (19). Finally, Bardet-Biedl syndrome causes a pigmentary retinopathy and photoreceptor degeneration that may have a cone–rod ERG phenotype because of associated central macular atrophy (see also Chapter 22).
Molecular Genetics of Cone–Rod Dystrophies
Like RP, cone–rod dystrophies can be inherited in an autosomal dominant, autosomal recessive, or X-linked recessive manner (Table 18.1). There are reports of potential associations with mitochondrial disease and cone dystrophies as well (20). Twenty or more genes have been identified to be associated with cone–rod dystrophies, while multiple other loci have been mapped.
Kohl et al. (21) determined that 50% of autosomal dominant cone–rod dystrophy was caused by mutations in the CRX, PRPH2, retinal guanylate cyclase (GUCY2D), guanylate cyclase activator 1A (GUCA1A), and PROM1 genes. Other genes playing a lesser role in the etiology of autosomal dominant cone–rod dystrophy include GUCA1A(GCAP1), GUCA1B(GCAP2), Rab3A-interacting molecule (RIMS1), AIPL1, UNC119, and PITPNM3 (3,22–25).
Genes associated with autosomal recessive cone– rod dystrophies are ABCA4, CNNM4, CACNA2D4, KCNV2, PDE6C, PDE6H, RPGRIP, RAXL1, CDHR1, ADAM9, SEMA4A, and RDH5 (14,19,26–28). Most of these genes have been identified in only isolated pedigrees. However, some groups suggest that in up to 35% of patients with autosomal recessive cone–rod dystrophy, the cause may be mutations in the ABCA4 gene (29). It is entirely possible that these groups are identifying patients who would otherwise have been characterized as having Stargardt disease or fundus flavimaculatus at different stages in the evolution of their clinical disease and not as having ABCA4-related cone–rod dystrophy. The distinction between these diseases given their clinical features and molecular genetics remains to be clarified.
The genes thus far identified to cause X-linked cone–rod dystrophy include RPGR, with the mutations occurring specifically in its ORF15 exon (CORDX1) (5). This exon seems to be a mutation hotspot, probably because the sequence is highly repetitive rendering it difficult to screen. The CACNA1F gene (CORDX3) was identified as a cause of cone–rod dystrophies in addition to X-linked congenital stationary night blindness (CSNB). One other locus for cone–rod dystrophy has been linked to the X chromosome but has not yet been identified (CORDX2).
Several genes associated with cone–rod dystrophies cause other retinal or macular dystrophies. Included among these are pattern dystrophies of the macula (peripherin/RDS), LCA (CRX, GUCY2D), RP (CRX, peripherin/RDS, ABCA4, RPGR), macular degeneration without full-field ERG abnormalities (RPGR) (30), CSNB (RDH5 and CACNA1F), complete achromatopsia (GNAT2), and Stargardt disease (ABCA4). Cone–rod dystrophies, like RP, demonstrate that mutations in the same gene can result in different clinical phenotypes previously considered to be entirely separate disease entities. See also Chapter 25 describing gene mutation and related protein function.
Choroideremia
Choroideremia is an X-linked disorder resulting in choroidal degeneration with subsequent loss of retinal function. It was first described in 1871 by Mauthner. The gene (CHM) was identified in 1992. Late-stage choroideremia has an unmistakable appearance of widespread loss of RPE, choriocapillaris, and larger choroidal vessels, with occasional patches of pigment and sparing of a small patch of RPE and retina in the macula. When choroideremia presents in childhood and adolescence, its symptoms and clinical appearance can be very similar to those of RP.
Clinical Presentation and Disease Course
Because of its X-linked inheritance, choroideremia typically presents in males in the second to third decade of life with the primary symptom of nyctalopia (31). On occasion, patients report symptoms as early as the first decade of life. Early in the course of the disease, nummular patches of RPE atrophy appear in the midperipheral retina. These areas become confluent with progressive loss of choriocapillaris initially and larger choroidal vessels subsequently (Fig. 18.3). The end stage results in bare sclera extending from the posterior pole to the periphery. In one study, Roberts et al. (32) found that 84% of patients younger than 60 years had 20/40 or better central vision, whereas 33% of patients older than 60 years had 20/200 or worse vision. Coussa et al. noted that visual acuity remained very stable until 50 years of age after which there was a rapid decline in central visual acuity. Thus, patients often maintain central vision until late in the course of disease but lose peripheral fields steadily from the time of diagnosis onward. As expected, the ERG shows progressive loss of retinal function throughout the disease course, eventually becoming nonrecordable in the late stages. The female carriers of mutations in the CHM gene, in this study, had good stable vision (33).
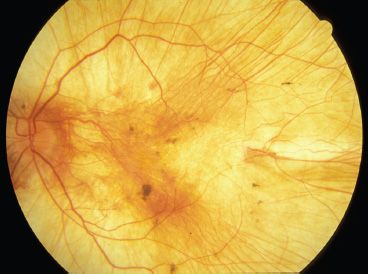
FIGURE 18.3 Choroideremia. Left fundus image of a 28-year-old man with choroideremia. He has severely constricted visual fields, and his central vision is 20/80 OD and finger-counting vision OS. Note the absence of RPE and choriocapillaris throughout the fundus. Large-caliber choroidal vessels are easily visible.
Female carriers of choroideremia demonstrate a spectrum of clinical manifestations ranging from essentially no findings to symptomatic loss of retinal function and apparent retinal degeneration (31). This spectrum likely is dependent on the degree of mosaicism and X inactivation. In general, carriers of mutations in the CHM gene demonstrate a moth-eaten appearance of RPE, with RPE clumping and atrophy. Many carriers also will demonstrate drusenoid deposition and peripapillary atrophy. However, some individuals will have a completely normal fundus. The degree of dysfunction on ancillary tests corresponds with the apparent clinical involvement.
Differential Diagnosis
Choroideremia can closely mimic the presentation of X-linked RP in its early stages. Both conditions have nearly identical clinical manifestations and symptoms and present at approximately the same age (i.e., second decade of life). Examination of the carriers (preferably, the mother of the patient) is helpful because the carrier state of X-linked RP can manifest with a tapetal macular sheen or with patches of retina characteristic of RP. Gyrate atrophy can also resemble choroideremia in its early stages; however, gyrate atrophy has an autosomal recessive inheritance pattern, hyperornithinemia, and a milder course than choroideremia.
Molecular Findings
The CHM gene was identified in 1993 by Sankila and further characterized by Seabra (34–36). The CHM gene codes for a Rab escort protein (REP-1) for the enzyme geranylgeranyl transferase, which is involved in adding a geranylgeranyl moiety to the end of Rab proteins (35–37). Nearly all disease-causing changes in the CHM gene were found to prevent translation or result in significant truncation of the protein product. There was one reported case of a missense mutation resulting in disease (38), although in a recent review of 57 families with choroideremia, missense mutations did not account for a significant fraction of disease-causing changes. Transitions and transversions in the CHM gene accounted for more than 40% of identified changes, small deletions (<5 base pairs [bp]) an additional 28% of the mutations, partial gene deletions 9%, and complete deletions 4% (39). At this time, the severity of the mutation does not appear to correlate with either disease severity or carrier state manifestations. Finally, the pathophysiology of disease from mutations in the CHM gene has yet to be elucidated. Syed examined the retina of an 88-year-old symptomatic carrier of the CHM gene and found changes suggesting that rod photoreceptors were the primary site of disease. In addition, REP-1 was localized to rods but not cones, further supporting rods as the primary site in disease pathophysiology. The choriocapillaris was atrophic in areas where the retina was severely degenerated but normal elsewhere (40).
CONGENITAL STATIONARY DISEASES
Congenital Stationary Night Blindness
CSNB includes a broad number of conditions marked by early-onset night blindness and a relatively stable clinical course (Table 18.2). The first description of CSNB was reported in 1838 by Cunier in a seven-generation French family (41). CSNB is subdivided into two categories based on the presence or absence of characteristic fundus changes. The forms of CSNB with characteristic findings display either white spots (fundus albipunctatus) or a greenish metallic sheen (Oguchi disease), whereas other forms of CSNB have minimal fundus changes. Most forms of CSNB have characteristic findings on dark adaptation and ERG and have a relatively good visual prognosis.
TABLE 18.2
Genes associated with CSNB and achromatopsia
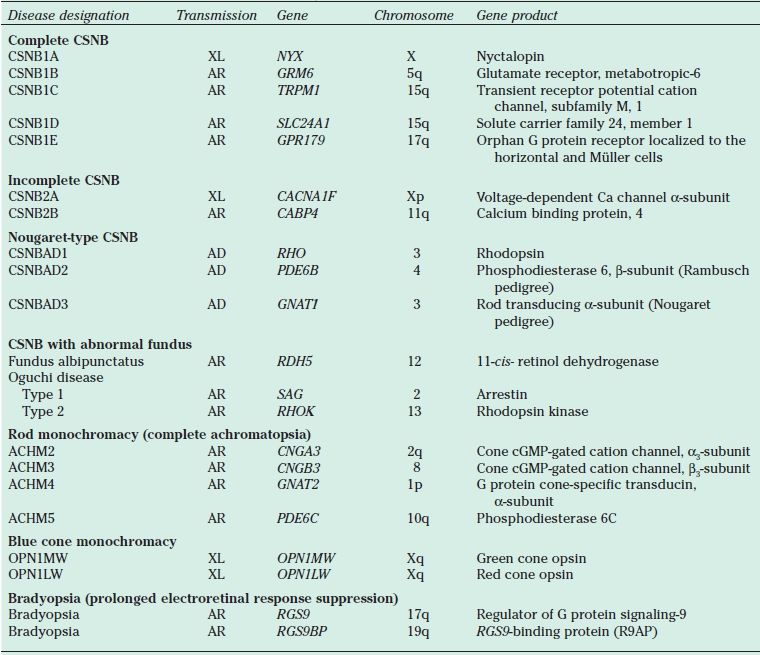
CSNB, congenital stationary night blindness; XL, X-linked; AR, autosomal recessive; AD, autosomal dominant; cGMP, cyclic guanosine monophosphate.
Normal Fundus Congenital Stationary Night Blindness
Complete and Incomplete Congenital Stationary Night Blindness
Both complete and incomplete CSNB (cCSNB and iCSNB) were first described as X-linked diseases. However, autosomal recessive patterns of inheritance have now been documented for both cCSNB and iCSNB. These forms of CSNB are characterized first and foremost by their ERG findings. Both cCSNB and iCSNB have the pathognomonic Schubert-Bornschein response (see below). These forms of CSNB are also characterized by significant refractive changes, nystagmus, and nyctalopia, although some reports indicate that many patients with X-linked CSNB may not present with nyctalopia (42). Best-corrected visual acuity usually is in the range from 20/40 to 20/50; however, vision can be better or worse.
Complete CSNB is characterized by severe progressive axial myopia and severe nyctalopia. Visual acuity may be relatively good, even 20/20 early in life (43). However, visual acuity subsequently is limited by myopia and astigmatic changes. Fundus findings are those consistent with high myopia, such as posterior staphyloma, scleral crescents, and tilted optic discs.
Incomplete CSNB demonstrates similar findings, although not all of these features need be present in each case. A study of 15 families with iCSNB reported that 72% of patients lacked at least one major feature, only around 40% of patients perceived nyctalopia, and 60% lacked nystagmus. Visual acuity ranged from 20/25 to 20/400 (43,44), with poorer visual acuities present in patients with nystagmus. Finally, some patients were hyperopic rather than myopic.
Both forms of CSNB demonstrate selective loss of the b-wave with an essentially normal a-wave, classically known as a Schubert-Bornschein response (45). This response differs from that in other conditions, such as X-linked retinoschisis or severe intraretinal ischemia, with a reduced b/a ratio in which the a-wave can be abnormal. ERG findings distinguish complete from incomplete CSNB (Fig. 18.4). iCSNB demonstrates relatively well-preserved rod function and abnormalities in the cone response, such as loss of the cone b-wave amplitude and a double-peaked 30-Hz flicker response. cCSNB demonstrates severely abnormal rod-isolated responses with normal or mildly subnormal cone responses (46).
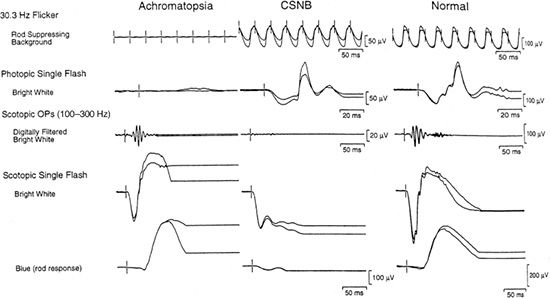
FIGURE 18.4 Full-field ERG of achromatopsia and cCSNB. ERGs of a 12-year-old girl with congenital achromatopsia, a 4-year-old patient with autosomal recessive cCSNB, and a normal for comparison. (From Weleber RG. Infantile and childhood retinal blindness: a molecular perspective [The Franceschetti Lecture]. Ophthalmic Genet 2002;23:71–97, with permission.)
The differential diagnosis for a patient with CSNB overlaps and includes RP, LCA, and achromatopsia. ERG findings coupled with relatively normal visual fields distinguish CSNB from these other conditions. Although almost pathognomonic for CSNB, the characteristic Schubert-Bornschein response can be seen in
Duchenne muscular dystrophy and has been reported in a family with autosomal dominant inherited negative ERG (47–49).
The gene for iCSNB was identified first in 1998. It is known as the CACNA1F gene and codes for the α-subunit of the calcium channel, which affects both the on pathway and, to a greater degree, the off pathway of the bipolar cells (44). Zeitz et al. (50) subsequently utilized the candidate gene approach to identify another cause of iCSNB, mutations in CABP4, which encodes a protein in the same family as that encoded by CACNA1F (50).
In 2000, the gene for cCSNB was identified as nyctalopin (NYX) (51). Mutations in nyctalopin are believed to disrupt the development of the ON pathways of the bipolar cells (43,44,51). Subsequently, the genes associated with autosomal recessive cCSNB, GRM6 and TRPM1, were identified by a candidate gene approach that targeted receptors and channels in the ON bipolar cells. A fourth gene causing cCSNB, SLC24A1, was isolated in one Pakistani family. A fifth gene, GPR179, was identified and reported by two separate groups as another uncommon cause of autosomal recessive cCSNB (52). The majority of mutations causing cCSNB and iCSNB have been identified in genes on the X chromosome.
Autosomal Dominant (Nougaret Type) Congenital Stationary Night Blindness
Autosomal dominant CSNB is characterized by a stable course, normal visual acuity, and normal visual fields. The fundus also appears normal, although some patients with rhodopsin-related CSNB may develop bone spicules in the midperiphery after middle age (14). ERG findings in the autosomal dominant form differ from X-linked CSNB in that a reduced response is present only under scotopic testing. This ERG pattern was initially distinguished from cCSNB and iCSNB by defining it as a Riggs-type ERG response. The rod-isolated responses in autosomal dominant CSNB are absent, whereas cone-isolated responses are normal. However, late in the course of disease, some patients with rhodopsin-related autosomal dominant CSNB can demonstrate mild cone dysfunction (14). In contrast to forms of CSNB associated with an abnormal fundus, the reduction in ERG response does not reverse with continued dark adaptation. The final dark-adapted threshold for rods is markedly elevated. The primary distinguishing features of autosomal dominant CSNB from milder forms of autosomal dominant RP are the stability of visual acuity, visual fields, and cone function.
Mutations in three genes have been associated with autosomal dominant CSNB. Mutations in codon 90, 94, or 292 of rhodopsin (RHO) are associated with a nonprogressive condition with normal vision and visual fields under mesopic or photopic situations and a reduced rod-isolated response as the major characteristic of nyctalopia (14). Evidence suggests that the associated changes in rhodopsin disrupt a critical salt bridge, thus changing the tertiary structure of the protein, and result in an opsin that is constitutively active. Consequently, the retina is desensitized and night blindness results (30). Finally, one change, p.Gly90Asp, has been associated with late onset of bone spicules (14). Changes in the genes for both the α-subunit of transducin (GNAT1) and the β-subunit of the rod cyclic guanosine mono-phosphate (cGMP) phosphodiesterase enzyme (PDE6B) have also been associated with autosomal dominant CSNB (41,53). GNAT1 has been implicated in the Nougaret pedigree from France first described by Cunier. PDE6B was found associated with the large Rambusch pedigree from Denmark (14,53,54).
Abnormal Fundus Congenital Stationary Night Blindness
Fundus Albipunctatus
Fundus albipunctatus was first described as a variant of retinitis punctata albescens by Lauber (55) in 1910. This condition is characterized by fine small white-yellow dots throughout the retina in association with a stationary, nonprogressive form of night blindness. In 1999, Yamamoto et al. (56) identified mutations in the gene encoding 11-cis-retinol dehydrogenase (RDH5) in patients with fundus albipunctatus. Although multiple mutations in the RDH5 gene have been associated with fundus albipunctatus, some families with a classic clinical appearance have not exhibited mutations in RDH5.
Clinical Features
The most characteristic feature of this condition is the presence of widespread white-yellow dots that are present in a radial pattern extending from the macula. These dots can fade as a patient reaches the fourth to fifth decade of life (14). This condition is characterized by subnormal rod-isolated responses with standard dark adaptation. Patients will frequently demonstrate an electronegative response with selective loss of the b-wave for the maximum combined response with standard dark adaptation. However, with extended dark adaptation, ERG responses improve and become essentially normal (Fig. 18.5) (14). Although this condition is classically considered to be a form of CSNB, evidence exists that, later in life, often the fourth to fifth decade (57), macular atrophy and an ERG phenotype consistent with cone–rod dystrophy can develop and persist even after prolonged dark adaptation (14,58). Many of these patients also demonstrate other macular changes, such as a bull’s-eye appearance. Typically, these changes occur in the fourth to fifth decades of life. Nakamura and Miyake identified a 9-year-old boy with atrophic foveal lesions and widespread white dots, who was a compound heterozygote for mutations in the RDH5 gene. This patient also demonstrated normalization of his ERG with no apparent residual cone dysfunction after 3 hours of dark adaptation (58). Thus, macular disease is not necessarily associated with cone dysfunction and may be present earlier in life, although these findings are more common in late adulthood.
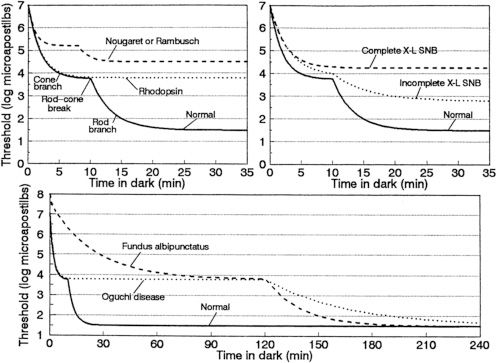
FIGURE 18.5 Dark-adaptation curves for patients with CSNB. Schematic dark-adaptation curves in normal individuals and in patients with stationary night blindness. In all three graphs, the y-axis is the minimal intensity of a perceptible spot of light (logarithmic scale, units are microapostilbs) at the time (in minutes) designated on the x-axis after exposure to light that bleaches 25% or more of the rhodopsin in each rod photoreceptor. The solid curve in each panel represents the normal dark-adaptation curve. The top left graph has the normal curve labeled with its components: the initial cone branch that ends at the rod–cone break and is followed by the cone branch. Top left: Dark-adaptation curves found in patients with the Nougaret or Rambusch forms of dominant stationary night blindness (similar forms of CSNB, represented by a single dashed line) and in a patient with the dominant rhodopsin mutation Ala292Glu causing stationary night blindness (dotted line, based on unpublished observations of E. L. Berson and M. Sandberg). Top right: Dark-adaptation curves from patients with complete (dashed line) or incomplete (dotted line) X-linked stationary night blindness. Bottom: Dark-adaptation curves characteristic of fundus albipunctatus (dashed line) and Oguchi disease (dotted line). The rod–cone break in both fundus albipunctatus and Oguchi disease has been arbitrarily placed at the 2-hour time point; the actual time point depends on the degree of light exposure preceding the evaluation of dark adaptation. (From Dryja TP. Molecular genetics of Oguchi disease, fundus albipunctatus and other forms of stationary night blindness: LVII Edward Jackson Memorial Lecture. Am J Ophthalmol 2000;130:547–563, with permission.)
The differential diagnosis for fundus albipunctatus includes other forms of flecked retinas such as Stargardt disease, pattern dystrophies, and retinitis punctata albescens. In many of these cases, the flecks are distinctly different. Nevertheless, there is some degree of phenotypic overlap among these conditions. Dark adaptation, fluorescein angiography, and ERG characteristics can help to distinguish these conditions. However, at one isolated visit in time, it may be difficult to separate these conditions even with clinical testing; therefore, repeated visits and the use of molecular genetic testing are useful.
Molecular Genetics
Fundus albipunctatus is caused by changes in the gene that codes for 11-cis-retinol dehydrogenase (RDH5) (56). This enzyme regenerates 11-cis-retinal in reti-noid metabolism and is believed to play a particularly critical role. However, to date, no other conditions besides fundus albipunctatus have been associated with mutations in this gene. Furthermore, Driessen et al. described members of two families with frame-shift mutations in the RDH5 gene that predicted essentially no function of the RDH5 gene. From their work, they concluded that RDH5 gene mutations would not be associated with more widespread retinal dystrophies other than fundus albipunctatus (59). Cideciyan et al. also evaluated the consequences of a null mutation in RDH5 on the visual cycle. Both of these groups ultimately concluded that other biochemical pathways besides 11-cis-retinol dehydrogenase exist for the production of 11-cis-retinal (60). The presence of these alternative pathways would explain, in part, the normalization of the ERG following prolonged dark adaptation. Cideciyan et al. also demonstrated that recovery of the cone response after a bleaching stimulus was unexpectedly slower than the recovery of the rod response. In fact, following a weak bleaching stimulus, the rate of recovery of rod sensitivity was independent of RDH5. Finally, some families with fundus albipunctatus have no abnormalities in the RDH5 gene, suggesting that there is some degree of genetic heterogeneity for fundus albipunctatus (61).
Oguchi Disease
This condition was first described by Oguchi (62) in 1907 as a cause of night vision loss. The majority of reported cases still originate in Japan. In 1995, this condition was found associated with mutations in the arrestin (SAG) gene and, shortly thereafter, with mutations in the rhodopsin kinase (RHOK) gene. It is commonly classified as a cause of CSNB with an abnormal fundus.
Clinical Features
Oguchi disease is characterized by the presence of a greenish metallic sheen in the fundus of a light-adapted patient. This sheen slowly fades with dark adaptation. After prolonged dark adaptation, the metallic sheen disappears completely but reappears following a few minutes of light adaptation; this is known as the Mizuo-Nakamura phenomenon. It has been hypothesized that excessive extracellular potassium in the retina due to overstimulation of rod photoreceptors plays a role in this clinical phenomenon (Fig. 18.6) (63). One group demonstrated by scanning laser ophthalmoscopy the formation and disappearance of fine dots in association with the Mizuo-Nakamura phenomenon and proposed these dots are related to the clinically observed metallic sheen and ERG findings (64). The ERG shows the presence of abnormal rod-isolated responses and an electronegative waveform on ERG testing. With prolonged dark adaptation of more than 4 hours, the ERG responses normalize and dark adaptation resumes normal rod thresholds. However, following a single flash stimulus, prolonged dark adaptation is once again required for a normal ERG response (Fig. 18.5) and for patients to see in a dark environment (14). Visual function under mesopic conditions is essentially normal with vision ranging from 20/25 to 20/50 (65,66). Beyond the Mizuo-Nakamura phenomenon, the fundus appearance in Oguchi disease is otherwise unremarkable, with no evidence of bone spicules, macular changes, or chorioretinal atrophy. However, some patients with arrestin mutations have family members with the symptoms and clinical signs of RP. Patients with arrestin-related RP also exhibit the Mizuo-Nakamura phenomenon early in their disease (66).
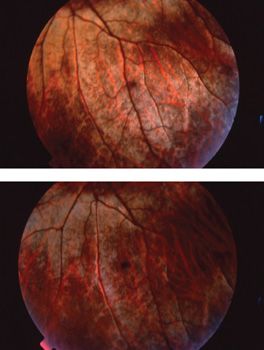
FIGURE 18.6 Mizuo phenomenon in Oguchi disease. (Courtesy of James Gilman, CRA, Ophthalmic Photography, Department of Ophthalmology, Moran Eye Center, Salt Lake City, UT.)
Molecular Genetics and Pathophysiology
Oguchi disease has been associated with mutations of the RHOK gene or the SAG gene (67,68). No other retinal dystrophies have been associated with changes in the rhodopsin kinase gene. However, a mutation in SAG, specifically c.1147delA, has also been associated with RP that manifests with decreased central vision, peripheral visual field constriction, midperipheral bone spicule pigmentation, and macular atrophy (69,70). The electrophysiologic and dark adaptation phenomena observed in Oguchi disease are directly related to the underlying molecular pathology of the disease. Rhodopsin activation occurs in the dark. Persistently activated rhodopsin (bleached rhodopsin) maintains rods in a desensitized state until all of the 11- trans– retinal, from previous light stimulation, is replaced with 11- cis-retinal. The inability to interrupt the phototransduction cascade due to defects in arrestin or rhodopsin kinase permits a minimal amount of active rhodopsin to maintain rods desensitized. It takes approximately 2 or more hours to completely regenerate bleached rhodopsin molecules (71); thus, the patient experiences a prolonged dark adaptation.
Achromatopsia
Achromatopsia is a congenital inability to discriminate color. It is a rare condition estimated to have a prevalence of 3 in 100,000. However, the prevalence of achromatopsia on the island of Pingelap in Micronesia is 4% to 10% due to a sharp reduction in the population after a typhoon that struck in the late 1700s. All affected individuals trace their lineage back to 1 of about 20 survivors, who was a carrier of the gene (72). Huddart first described in 1777 a patient with achromatopsia from a family with two affected brothers and three unaffected siblings. However, Daubeney is credited with the first overall description of achromatopsia in 1684 (73).
Achromatopsia is divided into two groups: complete achromatopsia (rod monochromacy) and incomplete achromatopsia (blue cone monochromacy). Complete achromatopsia is autosomal recessive and results in loss of all cone function, whereas incomplete achromatopsia is X-linked recessive and results in loss of red and green opsins only.
Clinical Characteristics
Both complete and incomplete achromatopsia are marked by poor color discrimination from birth, photosensitivity or photoaversion, decreased vision, and relative clinical stability. Nystagmus occurs in both forms of achromatopsia. However, by the time a patient presents for evaluation in late childhood or early adulthood, nystagmus may be barely noticeable. Patients with either form of achromatopsia tend to be myopic, sometimes with oblique astigmatism.
The two forms of achromatopsia are distinguished by the apparent inheritance pattern, although patients with incomplete achromatopsia can have better visual acuity (as good as 20/50 [43]); some color discrimination on certain clinical tests (74), such as the Sloan achromatopsia test (10); and macular changes with progression late in the course of the disease. In contrast, patients with complete achromatopsia tend to have visual acuity of 20/200 or worse.
Fundus findings in achromatopsia are relatively normal, with a reduced foveal light reflex in some cases. Some report macular atrophy in both forms of achromatopsia late in the course of the disease (43). Nathans et al. (75) have documented progression of disease in incomplete achromatopsia with macular atrophy. Nevertheless, both conditions have remarkably normal-appearing retinas and vessels. Electrophysiologic testing demonstrates loss of cone responses in the presence of essentially normal rod responses. In incomplete achromatopsia, minimal residual cone function can be detected with specialized testing, but under standard ERG testing, cone function typically is nonrecordable (Fig. 18.4).
The differential diagnosis for a child with decreased vision and nystagmus includes LCA, achromatopsia, CSNB, choroidal coloboma, peroxisomal disorders, optic nerve hypoplasia, and central nervous system causes. ERG characteristics and a patient’s presenting symptoms are useful in differentiating among conditions. A study by Lambert et al. (48) documented the misdiagnosis of LCA in 40% of patients. The most common misdiagnoses were CSNB, Joubert syndrome (retinal dystrophy with hypoplasia of the cerebellar vermis), RP, and achromatopsia. Other conditions that have been misdiagnosed as LCA include infantile Refsum disease, Zellweger syndrome, Senior-Loken syndrome (nephrophthisis and retinal dystrophy), cone–rod dystrophy, and Alström syndrome (47,48).
Molecular Genetics of Achromatopsia
Complete achromatopsia (rod monochromacy) is an autosomal recessive condition caused by defects in the cone photoreceptor cGMP-gated cation channel. The channel complex is composed of two α-subunits and two β-subunits. Mutations in the genes that code for each subunit have been implicated in the pathogenesis of rod monochromacy. The gene for the α-subunit (CNGA3) was the first gene identified and is located on chromosome 2q11 (76,77). Complete achromatopsia caused by changes in the CNGA3 gene is called complete achromatopsia type 2 and accounts for up to 25% of all patients with complete achromatopsia. The β-subunit is coded for by the gene CNGB3 and is believed to cause 40% to 50% of complete achromatopsia (76). CNGB3 is found on chromosome 8q21 and is called achromatopsia type 3. The inhabitants of the island of Pingelap carry a serine to phenylalanine missense change (p.Ser322Phe) that results in their disease (72). Another gene implicated in complete achromatopsia is GNAT2 located on chromosome 1p13. GNAT2 codes for the transducin α-subunit in cone photoreceptors. (GNAT1 codes for the α-subunit of transducin in rods.) Kohl identified five families with complete achromatopsia caused by homozygous changes in GNAT2 (43,78). Finally, PDE6C on chromosome 10q has also been reported in association with complete achromatopsia.
Incomplete achromatopsia (blue cone monochromacy) is caused by loss of the medium (green) and long (red) wavelength cone opsins encoded by genes that are found together on the X chromosome. Three mechanisms have been identified as causing incomplete achromatopsia. First, a locus control region (LCR) was identified 4.02 kb upstream from the start of the red opsin gene and more than 42 kb upstream from the start of the green opsin gene. The LCR region is responsible for unwinding the DNA prior to transcription regulated by the separate promoters of each respective opsin gene. A 579-bp deletion in the LCR has been identified in 18.2% of patients with incomplete achromatopsia studied; this mutation prevents transcription of either the red or green opsin genes (75). The second mechanism is related to unequal recombination of a red opsin gene with a mutant green opsin gene resulting in nonfunctional proteins. The final mechanism resulted from deletion of the green opsin gene with a missense mutation in the remaining red opsin gene (79). Thus, a deletion in a region up to 4,000 bp upstream from the 5’ initiation site of the red opsin gene can have an effect on the transcription and subsequent translation of a gene and result in incomplete achromatopsia.
Oligocone Trichromacy
Oligocone trichromacy was first reported by Van Lith in 1973 with additional patients subsequently being reported in the literature. This condition is extremely rare and related to achromatopsia. The clinical presentation of oligocone trichromacy differs from achromatopsia with normal or minimally affected color discrimination. Otherwise, patients can present with subnormal visual acuity, congenital nystagmus, a normal fundus, and photophobia. The ancillary studies also mirror achromatopsia with normal visual fields and an absence of cone function on ERG. The CNGB3 gene has been implicated, and Anderson et al. (80) suggest that this condition is a version of achromatopsia with preferential loss of peripheral cones.
Bradyopsia
Bradyopsia is a condition characterized by stationary cone dysfunction. It was first described in 1991 in a series of three unrelated Dutch patients (81). These patients presented with stationary subnormal vision, photophobia, and prolonged electroretinal response suppression. In 2004, Nishiguchi et al. identified mutations in the RGS9 or RGS9BP (R9AP) genes. The proteins encoded by these genes interact with trans-ducin to turn off the light-evoked response. Patients with bradyopsia present with mildly reduced visual acuity, photosensitivity, no dyschromatopsia, and normal or near normal color discrimination. Visual field testing shows mildly reduced general sensitivity possibly with more pronounced central loss of function, and full-field ERG testing demonstrates markedly abnormal cone responses. This condition has only been reported in a very limited number of subjects and is considered to be a stationary cone dysfunction condition (82).
MISCELLANEOUS GENERALIZED RETINAL DYSTROPHIES
Enhanced S-Cone Syndrome/Goldmann-Favre Disease
The enhanced S-cone syndrome (ESCS) was first described by Marmor et al. (83) based on specific electrophysiologic findings. Patients with ESCS presented with nyctalopia at an early age, cystoid changes in the macula, and an annular pigmentary retinopathy involving the region around and just outside the temporal vascular arcades (Fig. 18.7). Visual acuity could be as good as 20/20, but with the onset of cystoid changes of the macula, it could be as severe as 20/200. Color vision generally is unaffected, and the ERG shows a characteristic appearance. Rod-isolated responses are nonrecordable. However, the maximum combined response and photopic responses closely mirror each other in amplitude and waveform morphology. Spectral testing of the cones under photopic conditions demonstrates decreased function of long (L; red) and medium (M; green) wavelength sensitive cones and enhancement of short (S; blue) wavelength sensitive cones (83). Several patients with Goldmann-Favre syndrome also had these ERG characteristics. The gene for ESCS was identified as the NR2E3 gene in 2000 (84). This gene codes for a retinal orphan nuclear receptor, a transcription factor that plays a role in determining cone photoreceptor phenotype in embryogenesis. Without this receptor, cones remain as S cones without differentiating into L/M cones. However, some cones are still able to differentiate despite the loss of the NR2E3 gene product, which accounts for the normal color vision and relatively good visual acuity that can be exhibited by these patients. NR2E3 also is associated with a retinal degeneration resembling Goldmann-Favre syndrome described in Crypto-Jews from Portugal (85). These individuals demonstrate nyctalopia and constricted visual fields with a phenotype. Unfortunately, these patients were not studied with spectral ERGs to determine whether they also had ESCS (43).
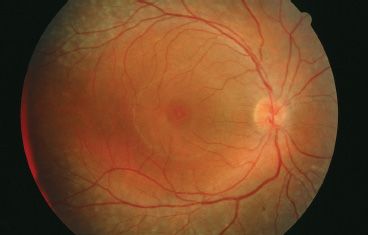
FIGURE 18.7 Fundus image of a patient with ESCS. Patient has cystic changes involving the fovea with midperipheral RPE changes characteristic of Goldmann-Favre/ESCS. This patient underwent ERG testing, including spectral ERGs, which demonstrated the characteristic findings of ESCS. (Courtesy of Richard G. Weleber, M.D.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


