Adapted from King RA, Hearing VJ, Creel DJ, et al. Albinism. In: Scriver CR, Beaudet AL, Sly WS, et al., eds. Metabolic and molecular bases of inherited disease, 8th ed. New York: McGraw-Hill, 2001:5587–5627.
Prevalence of Albinism
Albinism is a relatively common genetic condition. Its frequency varies considerably in the United States and around the world. The prevalence worldwide is about 1 in 10,000 to 1 in 20,000. In the United States, OCA2 is the most common in African Americans and Caucasians, with a prevalence of about 1 in 10,000 (1). OCA2 has a high prevalence in some Native American populations, such as the Hopi, Zuni (1), and Navajo. In the Navajo population, a unique deletion in the P gene is associated with an increased frequency of OCA2 (12). The prevalence of OCA1 is about 1 in 28,000 in African Americans and Caucasians. OCA3 occurs in about 1 in 8,500 in South Africa (1). HPS is rare overall but is relatively common in Puerto Rico, occurring in about 1 in 1,800 (13). HPS is also relatively common in an isolated village in the Swiss Alps (14).
Pathophysiology: Biochemistry of Melanin Synthesis
To understand the different defects causing albinism, it is helpful to be familiar with the melanin biosynthetic pathway. This pathway takes place within lysosome-related organelles known as melanosomes. Tyrosinase catalyzes the initial and rate-limiting step in melanin synthesis. In this step, the amino acid tyrosine is converted to dihydroxyphenylalanine (DOPA) and then to DOPAquinone. DOPAquinone represents a branch point, which can go in the direction of either pheomelanin (yellow/red) synthesis or eumelanin (brown/ black) synthesis. The pheomelanin pathway can occur in the presence of sulfhydryls. In the eumelanin pathway, DOPAchrome is formed and is converted to either 5,6-dihydroxyindole (DHI) or 5,6-dihydroxyindole-2-carboxylic acid (DHICA). Further processing of DHI results in DHI melanin (black) formation, whereas further processing of DHICA results in DHICA melanin (brown) formation. Tyrosinase is involved in both the DHI melanin and DHICA melanin pathways; however, in the DHICA melanin pathway, another protein, TYRP1, is also believed to play a role as a stabilizer of the melanogenic enzyme complex. Another enzyme, DHICA polymerase, is believed to catalyze the final step in DHICA melanin production (1).
As discussed in the section on Phenotypic Patterns of Albinism, the different albinism phenotypes result from corresponding defects at different levels affecting the above pathway, including (i) enzymatic steps in the pathway, (ii) proteins important for the melanosomal organelle and transport, and (iii) proteins important for multiple organelles, including melanosomes.
Clinical Symptoms and Signs
Normally, melanin accumulates in the retinal pigment epithelium during the early embryonic stages. This is important for appropriate routing of optic nerve fibers and for eventual visual function. The ocular findings of albinism are characteristic. Decreased melanin causes iris translucency and retinal hypopigmentation (1–3,15) (Figs. 17.1 and 17.2).
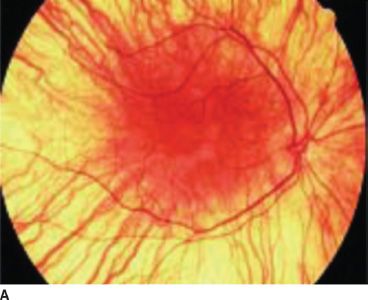
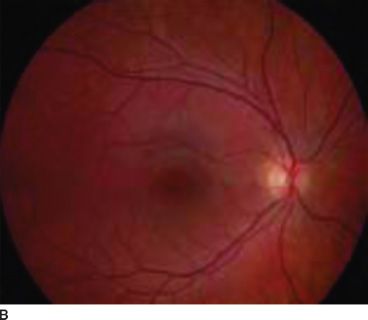
FIGURE 17.1 Hypopigmented fundus in albinism (A) and fundus of a normal eye (B). (Reprinted from Gronskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis 2007;2:43, with permission.)

FIGURE 17.2 Eyes from an OCA1A patient. Note pinkish, translucent irises. (Reprinted from Gronskov K, Ek J, Brondum-Nielsen K. Oculocutaneous albinism. Orphanet J Rare Dis 2007;2:43, with permission.)
Decreased visual acuity occurs in albinism and is due to foveal hypoplasia. Visual acuity may range from 20/40 to 20/400. Color vision is normal. A very characteristic finding in albinism is the misrouting of optic nerve fibers. Normally, fibers from the nasal retina cross at the optic chiasm and end at the lateral geniculate nucleus on the contralateral side. Temporal fibers do not cross over. In albinism, the temporal fibers inappropriately cross at the chiasm. The result is that the proportion of crossed fibers is increased. Misrouting of the optic nerve fibers can sometimes be detected by visual evoked potential (VEP) (1,2). Other ocular features of albinism include nystagmus, strabismus, and abnormal stereoscopic vision (1,2).
Phenotypic Patterns of Albinism
OCA1
Clinical Symptoms and Signs: Patients with OCA1 (“tyrosinase related”) are characteristically born with severe hypopigmentation of the skin and hair (1–3). In OCA1A, which is associated with complete absence of tyrosinase activity, the hypopigmentation does not significantly change over time. No freckles or other pigmented skin lesions develop. In contrast, OCA1B is associated with varying levels of residual tyrosinase activity. As in OCA1A, there is severe hypopigmentation of the skin and hair at birth; however, patients with OCA1B tend to develop some pigmentation over time. They may develop freckles and some tanning, and the hair may develop a brown or yellow coloration. The ocular features of OCA1B are generally milder than those in OCA1A. The varying levels of tyrosinase activity in OCA1B cause corresponding differences in phenotype. Some clinical entities, such as yellow albinism, temperature-sensitive albinism, and platinum or mixed or minimal pigment albinism, now are considered to be phenotypes within the OCA1B spectrum (1).
Genetics: OCA1 is caused by mutations in the TYR gene, which encodes for tyrosinase (4).
OCA2
Clinical Symptoms and Signs: As mentioned previously, OCA2 (“tyrosinase-positive”) is the most common form of albinism in the United States and worldwide. In contrast to OCA1, some pigmentation usually is present at birth. Similar to OCA1B, patients may undergo some tanning and can develop freckles. OCA2 sometimes is referred to as tyrosinase-positive OCA because of the normal tyrosinase activity (1).
Genetics: The defect in these patients is in the P gene, which encodes for a protein thought to be involved in maintenance of acidic pH in the melanosomes (16). The P-gene product also may play a role in intracellular transport of tyrosinase and possibly a minor role in transport of TYRP1 (17). There is marked clinical variability within the spectrum of OCA2. In Africans and African Americans, a classic phenotype has been described consisting of congenitally yellow hair and a creamy white skin color. The irides are usually blue-gray, hazel, or tan (1). Some African American and African patients have a phenotype called brown OCA, which also is due to mutations in the P gene (1,18,19).
The P gene maps to chromosome 15 within the Prader-Willi syndrome and Angelman syndrome region. Prader-Willi and Angelman syndrome are complex conditions with mental retardation (20,21). The majority of these patients have deletion of 15q11–q13, and a substantial proportion have hypopigmentation related to the P gene. The mouse locus for the P gene is “pink-eyed dilution” (p) (16).
OCA3
Clinical Symptoms and Signs: OCA3 has been described in a number of patients in South Africa. These individuals tend to have reddish hair color and hazel or brown eyes; the skin color is described as reddish brown (Fig. 17.3). The ocular features are mild. Misrouting of optic fibers has not been described. The entity known as rufous albinism is now considered to be OCA3 (1,22).

FIGURE 17.3 Two South African siblings (front) with atypical OCA. Mutation testing showed compound heterozygosity for mutations in the TYRP1 gene but also a 2.7-kb deletion of the P gene. In addition, an unrelated woman (back right) reportedly has rufous OCA. Mutation testing showed compound heterozygosity for mutations in the TYRP1 gene. (Reprinted from Manga P, Kromberg JG, Box NF, et al. Rufous oculocutaneous albinism in southern African Blacks is caused by mutations in the TYRP1 gene. Am J Hum Genet 1997;61:1095–1101, with permission.) Hartnett 9781451151404-ch17.
Genetics: The molecular basis of OCA3 is mutation in the TYRP1 gene (23). The encoded protein TYRP1 is involved in the eumelanin pathway to form DHICA melanin (brown). TYRP1 is thought to stabilize the melanogenic enzyme complex. The corresponding mouse locus for TYRP1 is “brown” (b). TYPR1 is involved in eumelanin synthesis and is believed to have other roles such as stabilizing tyrosinase, affecting tyrosinase activity, and maintenance of melanosomal structure and proliferation (23).
OCA4
OCA4 is associated with a similar phenotype to that of OCA2 (8). The gene involved in OCA4 encodes for a membrane-associated transporter protein. The mouse homolog is underwhite (uw) (8).
Ocular Albinism (Nettleship-Falls Type)
Clinical Symptoms and Signs: The phenotype of OA1 is similar from an ocular standpoint to OCA. However, the skin and hair pigmentation are normal, although scattered hypopigmentation has been observed in some African American patients. Giant melanosomes are found in the melanocytes of OA1 patients.
Genetics: OA1 is inherited in an X-linked recessive fashion; therefore, males are predominantly affected (1). However, carrier females may have a mosaic pattern of pigmentation of the fundus due to lyonization (Figs. 17.4 and 17.5) (24). The gene maps to Xp22 and is designated GPC143 (11). The corresponding mouse locus for OA1 is “OA1” (moa) (25).
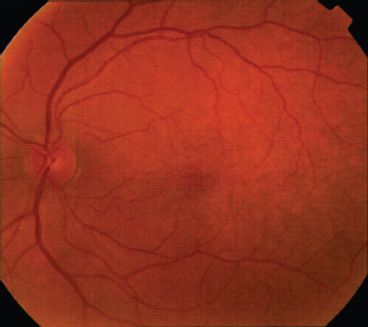
FIGURE 17.4 Left macula of OA1 carrier female. Note mottling and patchy hypopigmentation of the retinal pigment epithelium. (Reprinted from Costa DLL, Huang SJ, Donsoff IM, et al. X-linked ocular albinism: fundus of a heterozygous female retina. J Retinal Vitreous Dis 2003;23:410–411, with permission.)

FIGURE 17.5 Composite photographs of the right eye of an OA1 carrier female. Note “mud-splattered” hyperpigmented streaks. (Reprinted from Costa DLL, Huang SJ, Donsoff IM, et al. X-linked ocular albinism: fundus of a heterozygous female retina. J Retinal Vitreous Dis 2003;23:410–411, with permission.)
Hermansky-Pudlak Syndrome
Clinical Symptoms and Signs: HPS is a multisystem disorder, one component of which is OCA (1,9). The other major components are bleeding problems due to absent platelet delta granules (Fig. 17.6), ceroid storage in multiple tissues, and increased risk for granulomatous colitis and pulmonary fibrosis. The degree of hypopigmentation of the skin and hair is quite variable. Visual acuity is decreased, and the other typical ocular features of albinism are present. Normally, when platelets are stimulated, platelet delta granules release compounds that lead to the secondary aggregation response. The lack of platelet delta granules in HPS prevents normal secondary aggregation (1,9).
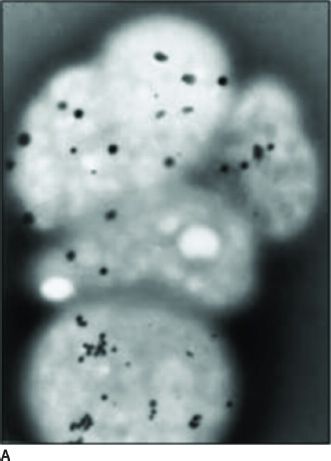
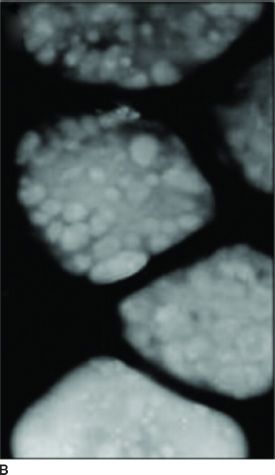
FIGURE 17.6 Absent platelet delta bodies. A: Electron micrographs of normal platelets containing delta granules (black dots). B: Absent delta granules in MPS platelets (Reprinted from Huizing M, Helip-Wooley A, Westbroek W, et al. Disorders of lysosome-related organelle biogenesis: clinical and molecular genetics. Annu Rev Genomics Hum Genet 2008;9:359–386, with permission.)
Genetics: HPS is genetically heterogeneous with at least eight known subtypes. The genes associated with HPS encode for proteins important for the formation of vesicles such as melanosomes, lysosomes, and platelet dense bodies (1,9).
Chediak-Higashi Syndrome
Clinical Symptoms and Signs: CHS patients have hypopigmentation of the skin, hair, and eyes. The hair tends to have a metallic silver-gray sheen (Fig. 17.7). The skin may have a white creamy appearance. There is variable pigmentation in the iris and variable presence of nystagmus and photophobia. Immunologic problems are characteristic and include defects in natural killer cell activity and defective chemotaxis. Recurrent pyogenic bacterial infections are common. Platelet dense bodies are abnormal, and there is a tendency to bleeding. A large proportion of patients develop lymphoproliferative infiltration of the bone marrow and reticuloendothelial system. Hematopoietic stem cell transplant is curative for the hematologic and immunologic aspects of CHS. Multiple neurologic problems develop over time (9,26).
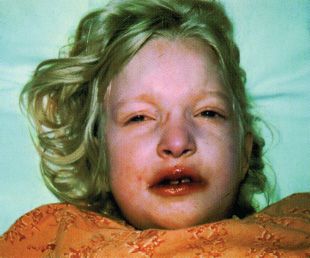
FIGURE 17.7 Patient with CHS. (Reprinted from Weinberg S, Prose N, Kristal L. Color atlas of pediatric dermatology, 3rd ed. New York: McGraw-Hill, 1998:237, with permission.)
Genetics: The gene for Chediak-Higashi is LYST, the product of which is thought to be involved in regulation of lysosomal trafficking; the corresponding mouse locus is “beige” (bg). CHS is considered to be a disorder of lysosomes and lysosome-related organelles (10).
Diagnosis of Albinism
Clinical diagnosis of albinism is based on oculocutaneous or ocular hypopigmentation along with the characteristic ocular abnormalities in albinism. If necessary, VEP studies should be done to detect misrouting. Other aspects of evaluation include general ophthalmologic assessment for decreased visual acuity, nystagmus, strabismus, foveal hypoplasia, and abnormalities in depth perception (1).
An important clinical distinction between OCA1 and OCA2 is the presence of visible pigmentation at birth in OCA2, whereas in both types of OCA1 the newborn appears to have white skin and hair. For OA1, electron microscopic evaluation of skin or hair bulb melanocytes can be performed to detect macromelanosomes to help in the diagnosis of this condition (1). DNA testing is clinically available for OCAs 1 to 4 and for OA1 (see Genetests in Resources section).
The diagnosis of HPS is based on the presence of OCA in combination with the other characteristic features of HPS. The absence of platelet dense bodies on electron microscopy is diagnostic of HPS. The diagnosis of CHS is based mainly on the clinical features (1). Huge lysosomes and cytoplasmic granules are important diagnostic findings for CHS. DNA testing is clinically available for HPS and for CHS (see Genetests in Resources section).
Management
Ongoing care with the ophthalmologist and dermatologist is important. Ocular management is through low-vision rehabilitation and filters (see Chapter 33). However, future treatments may be developed to correct the genetic defect, such as through gene therapy (see Chapter 21).
Dermatology is important mainly to advise the patient on measures to take for protection against the sun.
Although a diagnosis of albinism is evident in most cases, it is useful to refer the patient to a genetics clinic to help determine the type of albinism and to provide appropriate genetic counseling, pedigree analysis, and deoxyribonucleic acid (DNA) testing if warranted. Genetic evaluation can be helpful in ruling out or diagnosing complex syndromes with albinism, such as HPS or CHS. If either is diagnosed, other consultations are needed, depending on complications and monitoring. The care for these patients may be coordinated by the clinical geneticist or other physicians experienced in the management of rare diseases.
The lysosomes are cytoplasmic organelles that serve as the cellular compartments where macromolecules are degraded into their individual constituents. To perform this function, lysosomes contain hydrolytic enzymes to carry out the digestion of these macromolecules. Numerous diseases due to deficiency of various lysosomal enzymes have been described. The result of individual defects is the accumulation of the macromolecule substrate for which the enzyme was responsible for degrading. The clinical phenotype is determined by the specific substrate and the effect of its accumulation on various tissues. From a clinical standpoint, a consistent characteristic of most lysosomal storage diseases is developmental regression. This is caused by the ongoing damage due to accumulation of the undegraded substrate.
Most of the lysosomal storage diseases, except for the X-linked Hunter syndrome and Fabry disease, are autosomal recessive.
The major groups of macromolecules degraded in the lysosomes include glycoproteins, glycolipids, and glycosaminoglycans (mucopolysaccharides).
Glycoproteins are compounds with varying proportions of carbohydrate molecules, such as fucose, man-nose, sialic acid, and galactose attached to proteins. Disorders of glycoprotein breakdown include fucosidosis, mannosidosis, aspartylglucosaminuria, sialidosis, and galactosialidosis (27).
A variety of complex lipid-containing compounds are degraded in the lysosomes. Sphingomyelin is a phospholipid and contains ceramide attached to phosphocholine and a long chain fatty acid. The term “glycolipid” refers to compounds with a glucose or galactosyl group attached to ceramide. Defects in the degradation of the above compounds include Niemann-Pick disease (NPD), Tay-Sachs disease, Sandhoff disease, GM1 gangliosidosis, Krabbe disease, Gaucher disease, and others. Farber disease is due to a defect in the degradation of ceramide, which is a core component of phospholipids and glycolipids (27).
Glycosaminoglycans (mucopolysaccharides) consist of polymers of hexuronic acid attached to amino sugars. The glycosaminoglycans are important components of connective tissue ground substance. Disorders in this category are called mucopolysaccharidoses and include Hurler syndrome, Hunter syndrome, Sanfilippo syndrome, Scheie syndrome, Morquio syndrome, Maroteaux-Lamy syndrome, and Sly syndrome (27).
Retinal abnormalities are common in lysosomal storage disease. For example, cherry-red spot in the macula is a consistent feature of several of the disorders of glycolipid and glycoprotein degradation (Table 17.2). Retinal abnormalities along with other ocular problems such as corneal clouding and glaucoma are common in the mucopolysaccharidoses.
TABLE 17.2
Selected lysosomal storage disorders consistently associated with cherry-red spot macula

PPCA, protective protein/cathepsin A.
Defects in Glycoprotein Metabolism
Sialidosis
Symptoms and Signs: Sialidosis is an autosomal recessive disorder of glycoprotein degradation. Clinically, this disorder can be classified into a less severe form, type 1, and a more severe phenotype, type 2. Type 2 is itself divided into congenital, infantile, and juvenile subtypes. A cherry-red spot macula occurs commonly in sialidosis, especially in type 1, and may appear similar to that seen with artery occlusions (28,29).
Type 1 sialidosis has been referred to as cherry-red spot myoclonus syndrome. Patients with this disorder may have normal development for the first two to three decades of life and then manifest visual and neurologic problems. Night blindness and problems with color vision are relatively common. Funduscopic evaluation demonstrates a cherry-red spot in the macula (30). Other ocular features may include nystagmus or opacities of the lenses or corneas. Neurologic problems occur including seizures, ataxia, and dysarthria (28,31).
Type 2 sialidosis has an earlier onset and includes a congenital form, which can present with hydrops. In contrast to type 1, somatic features are characteristic. These may include enlarged liver and spleen, ascites, abnormal facies, and a radiographic pattern known as dysostosis multiplex. A cherry-red spot is seen in the majority, but not all, of type 2 patients (28).
Diagnosis: The defect in sialidosis is neuraminidase deficiency. The diagnosis is based on a characteristic oligosaccharide pattern in the urine and assay of the neuraminidase enzyme (28).
Defects in Metabolism of Sphingomyelin, Gangliosides, and Other Complex Lipids
Niemann-Pick Disease Types A and B
Symptoms and Signs: Type A NPD is an autosomal recessive disorder caused by deficiency of acid sphingomyelinase. Normally, this enzyme is involved in the lysosomal degradation of sphingomyelin. Patients present in early infancy with hepatosplenomegaly, hypotonia, and weakness. Developmental delay is evident by about 6 months of age. Development regresses, and patients become rigid and spastic and lose contact with their environment. During the first 12 to 24 months of life, many patients are found to have cherry-red spots in their macula and abnormal electroretinograms (ERGs). Some patients develop a gray granular appearance to the macula referred to as the macular halo syndrome (32,33).
Type B NPD also is due to acid sphingomyelinase deficiency; however, the clinical spectrum is much milder. Clinical presentation is later and, in some patients, splenomegaly is not apparent until adulthood. Respiratory problems due to alveolar infiltration may occur at 15 to 20 years of age. The central nervous system is usually spared. A few type B patients have cherry-red spots in the macula.
Diagnosis: The diagnosis of Niemann-Pick types A and B is suspected based on the clinical features and bone marrow findings (NPD “foam” cells). Confirmatory enzyme studies for acid sphingomyelinase, such as from peripheral leukocytes, cultured fibroblasts, and lymphoblasts, are necessary for diagnosis (32).
Disorders of Ganglioside Degradation: GM1 Gangliosidosis
Symptoms and Signs: GM1 gangliosidosis is an autosomal recessive neurodegenerative disease due to deficiency of β-galactosidase. The clinical presentation is variable and includes infantile, late infantile/juvenile, and adult/chronic forms. Most patients present in early infancy, usually with arrested development several months after birth. This is followed by developmental regression. Exaggerated startle response and seizure are common. Rigidity, spasticity, and seizures are frequent. Dysmorphic features, skeletal dysplasia, and hepatomegaly are characteristic, and patients eventually regress to a vegetative state. Ophthalmologic evaluation reveals optic atrophy, corneal clouding, retinal edema, and macular cherry-red spot. In the late infantile/juvenile form, the onset is later, but developmental regression leads to early death. There are dysmorphic facial features and the skeletal pattern of dysostosis multiplex. Cherry-red spots in the macula occur in the late infantile form but not as consistently as in the early infantile presentation (34,35).
Diagnosis: Enzyme assay for β-galactosidase is performed for diagnostic confirmation (34,35).
Disorders of Ganglioside Degradation: GM2 Gangliosidosis (Tay-Sachs Disease)
Tay-Sachs disease is an autosomal recessive condition due to a deficiency of β-hexosaminidase A (Hex A), which normally participates in the hydrolysis of gan-glioside GM2. Hex A deficiency results in the accumulation of ganglioside GM2 in the lysosomes of the nervous system causing progressive tissue damage. There is marked variability in the clinical patterns, ranging from the classic infantile disease to later onset and even an adult form of the condition.
Symptoms and Signs: In the acute infantile presentations, patients develop some degree of motor weakness by age 3 to 5 months. Exaggerated startle responses, seizures, and rapid developmental regression occur. A cherry-red spot is seen on funduscopic examination (Fig. 17.8). ERG is normal; however, visual evoked responses are abnormal early on. The disease progresses rapidly after about 8 to 10 months of age as the infant becomes less responsive to the environment. Ultimately, Tay-Sachs patients become vegetative and usually do not survive beyond age 3 to 5 years (36).
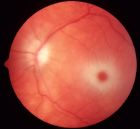
FIGURE 17.8 Color fundus image of a left eye from a patient with Tay Sach’s disease (GM2 gangliosidosis or hexosaminidase A deficiency) showing a cherry red spot. The accumulation of gangliosides or sphingolipids within the retinal ganglion cells produce a white appearance surrounding the fovea, which lacks ganglion cells and can transmit a natural red appearance from the underlying choroid. (Courtesy of Elias Traboulsi, MD, Cole Eye Center, US.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


