FIGURE 16.1 Bilateral nasal tilted disc syndrome, associated with inferonasal staphyloma in a 20-year-old patient. Vision is 20/20 with a corrected astigmatism. Right eye (A), left eye (B).
1. Subnormal vision
2. Myopic astigmatism with oblique cylinders
3. Transposition (tilting) of the optic disc downward and nasally
4. Inferior or inferonasal crescent (congenital conus)
5. Nasal or inferonasal ectasia associated with thinning of the pigment epithelium and choroid
6. (Bilateral) temporal loss of visual field (5–8)
Many reports (6,9–12) stated that more women than men had tilted discs. Although many studies reported a high frequency of bilateral involvement (9–12), there are a number of cases that remain misdiagnosed. Vongphanit et al. (4) found that the tilted disc appearance was bilateral in 37.5% of cases, whereas Riise (10) included that “slighter cases… which would have been described as normal in the usual ophthalmoscopy” occur bilaterally 74% of the time.
Prevalence
In the majority of cases (80%), the tilted disc syndrome is bilateral. There have been very few past reports of the prevalence of tilted discs. In a report from a Norwegian practice series (Hamar) of 300 participants aged 7 years or older, the tilted disc syndrome was found in five (1.7%) (10). An identical figure was found for tilted discs in an Italian community (Casteldacchia) of 300 participants (13). In a German report from 1922 that included tilted disc illustrations, von Szily (14) reported that 3.4% of 32,500 eyes from a clinic had a nontemporal (“heterotypical”) distribution of peripapillary atrophy (PPA) described by some authors as the quintessential feature of the tilted discs (10,15).
The prevalence in an Australian population was 1.6% and is comparable to the rate of 1.7% reported by both Giuffre (11) and Riise (10). Given that the tilted disc appearance varies considerably, even between eyes within an individual (10), the use of dysversion alone may lead to an underestimation of its prevalence and bilaterality. Dysversion of the optic nerve head is the complete form of the tilted disc syndrome with all the clinical and functional alterations. It has also been referred to as segmental hypoplasia and is characterized by central retinal vessels emerging temporal to the vertical midline of the disc and being directed nasally or the nerve head tilting in a vertical direction resulting in a downward or oblique tilting of the discs with the blood vessels emerging at the superior or inferior disc rim.
Tilted optic discs occur in about 4 out of 1,000 non– highly myopic eyes of adult Chinese (16). As in Caucasians (1,3), tilted discs were associated with moderate myopia, astigmatism, inferiorly located beta zone of PPA, decreased visual acuity, and visual field defects. One may infer that a visual field defect in an eye with a tilted disc is not proof of an acquired optic nerve disease such as glaucoma.
Etiology
It is thought that the spectrum of signs present in any given eye is dependent on the severity of the embryonic optic fissure malclosure at the 6-week stage of gestation (8). The 6th week shows the incipient differentiation of the inner layer of the optic cup into a sensory retina, the formation of the secondary vitreous, the transformation of the posterior cells of the lens vesicle into primary lens fibers, the development of the periocular vasculature, and the appearance of the first eyelid folds and of the anlage of the nasolacrimal duct.
The spectrum ranges from a minimal defect (tilted disc) to more severe fundus involvement (i.e., inferonasal ectasia, frank coloboma) (5).
In some opinions, this assumption does not explain adequately the origin of all ocular anomalies found in the syndrome, like the refractive errors (myopia and hypermetropia, corneal astigmatism) and the various types of visual field defects. Fewer retinal nerve fibers than normal enter the defective side of the disc (15), whereas the opposite side presents fiber crowding, which may give an erroneous impression of edema. Therefore, it has been suggested that atrophy of the ganglion cell fibers of the inferonasal retina might occur following an anomalous closure of the embryonic cleft. This in turn would explain the hypoplasia of the inferonasal quadrant, the inferior crescent (lack of fibers in the inferior optic disc), and the anomalous appearance of the optic nerve head and of the retinal vessels (shifting of the disc upward due to the imbalance between the ganglion cell fibers coming from the superior and inferior retina) (11). Furthermore, this accounts for the upper temporal visual field defects mostly found in the syndrome.
The heredity of the condition is not yet established, perhaps because of the variability in the phenotype in family members of affected individuals; hence the lack of reports of the familial nature of the syndrome. Single clinical features like inferior crescent or transposition of the optic disc have been reported as associated with various inheritance patterns (10). On the other hand, Riise (10) believes that the tilted disc syndrome can be familial, with a polymeric mode of inheritance similar to that of refraction anomalies. Bottoni et al. (17) presented a family with dominant penetrance of tilted disc. The tilted disc syndrome has also been reported in patients with X-linked congenital stationary blindness (14). The tilted disc syndrome is believed to be nonprogressive (5).
Clinical Appearance
The most frequent orientation of tilted discs is inferonasal (60%) (Fig. 16.1), thus tilting to the nasal side of the vertical meridian; however, inferotemporal tilting has also been reported (28%) (5). The appearance of tilting arises from the pseudorotation of the superior pole of the optic disc (5), angulation of the optic cup axis inferonasally, and elevation of the superotemporal neuroretinal rim (6). The optic disc configuration of tilted discs can sometimes be confused with optic disc swelling, papilledema (6,8), or presumed optic disc drusen (9), as the elevated retinal nerve fiber layer appearance may cause blurring of the disc margin and visual field changes may also be present. Although inferonasal tilting is most frequent, inferior tilting is the most severe (4).
Best-corrected visual acuity is reduced in eyes with tilted discs. This is believed to be due to the Stiles-Crawford effect as the photoreceptors stand obliquely in relation to the visual axis (10). Several reports (6,8–10,12,18,19) revealed that a high proportion of people with tilted discs had myopia and astigmatism with the plus axis parallel to the fundus ectasia in the inferior quadrant. Corneal topography studies indicate that an irregular curvature contributes to the associated astigmatism—thus a corneal origin of the astigmatism (20). Vongphanit et al. (4) have quantified this relationship, showing a strong association between the tilted disc appearance and the level of either astigmatism (93.5%) or spherical refractive error; myopia was present in 62% of subjects with tilted disc syndrome compared to individuals with normal optic discs. Oblique astigmatism was also present in over 50% of the cases.
Reduced visual acuity in association with bitemporal depression of the visual field and that is typically incomplete corresponding to the inferonasal optic nerve fiber loss (Fig. 16.2) can suggest a lesion near the optic chiasm. Recognition of the fundus abnormalities, possible improvement of the field defect with proper refractive correction (10), and lack of the vertical boundary at the midline of the visual field should obviate the need for a major neurologic workup.
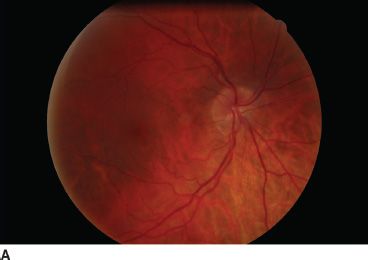
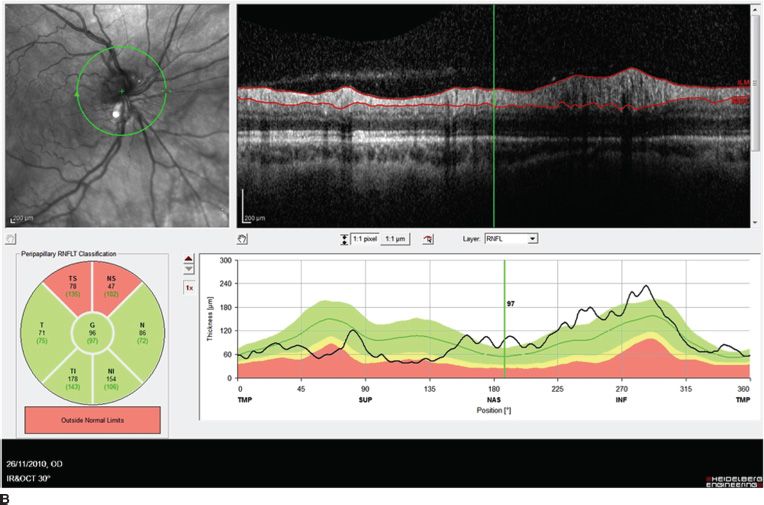
FIGURE 16.2 Nasal tilted disc syndrome, fundus appearance (A), and OCT scan showing optic nerve fiber layer swelling on the nasal inferior and superior portion of the disc (B).
Many field defects have been described in association with the tilted disc syndrome. The most frequent sites are superotemporal, temporal, and superior (6,9,12), but nasal, inferior, and paracentral defects have also been reported. Some studies have reported that the majority of patients with tilted discs had a visual field defect, whereas other studies have indicated that field defects were not consistently found (15,19). In some studies, visual field defects were associated with myopia over 6 diopters (8,21). Repeat perimetry after addition of a -4 lens often eliminated the visual field abnormality, confirming the refractive nature of this defect (6).
Visual field defects associated with “scooping out” of the optic disc have been misdiagnosed as glaucomatous (Fig. 16.3) (1,8). Bitemporal hemianopia associated with tilted discs may also be confused with field defects resulting from chiasmal compression leading to unnecessary neuroradiologic investigations (1,10).
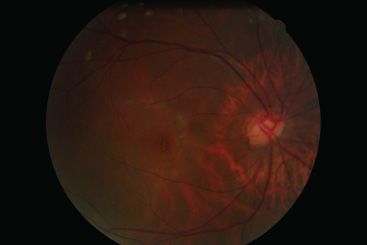
FIGURE 16.3 Tilted optic disc with marked central excavation.
Certain features of the field defect help distinguish defects associated with the tilted disc syndrome from other pathology. The defect tends to be relative in nature (9,10) and nonprogressive (6,8) and may resolve with a larger test stimulus or after optical correction (9,10). It is usually surrounded by a normal peripheral visual field and frequently crosses the vertical meridian of the visual field (6,9).
Although not frequently associated with severe visual impairment, tilted discs may be associated with choroidal neovascularization (7,11,18,19), chorioretinal atrophy (18), and neurosensory macular detachment (2). Posterior staphyloma is commonly associated (11,13) and may affect vision due to choroidal neovas-cularization and chorioretinal atrophy (5). Anomalies at the junction of the staphyloma may cause serous macular detachments (Fig. 16.4).
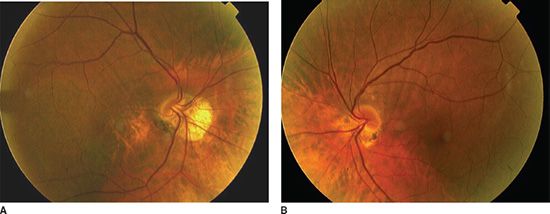
FIGURE 16.4 Aspect of Figure 16.1. Patient 10 years later showing marked pigment epithelial changes in the area of the inferior staphyloma and peripapillary inferonasal atrophy. Right eye (A), left eye (B).
Giuffre (11) and Vongphanit (4) in their respective case series described inferonasal pigmentary accumulation in 11% of older patients with severe disc tilting. Eyes with pigmentary accumulation were more likely to be myopic and to have poorer visual acuity than eyes without pigmentary accumulation. Although pigmentary accumulation could resemble myopic lacquer cracks (10), when seen in eyes with tilted discs, it was located in the inferonasal retina and had a coarse appearance presenting a circular pattern around the disc (22).
Conclusion
In children, detection of tilted discs may prompt refractive measurement and correction to exclude corneal astigmatism and prevent amblyopia.
When tilted disc syndrome is accompanied by a visual field defect that respects the vertical meridian, neuroimaging is mandatory.
Introduction
Optic nerve head drusen (ONHD) are a common, benign, congenital anomaly of the optic nerve, which can rarely lead to decreased visual acuity (23,24). ONHD are laminated calcified hyaline bodies arising from the prelaminar portion of the optic nerve (23,25–27). They were first described clinically in 1868 by Liebrich (28) and histopathologically by Muller in 1858 (29). They occur almost exclusively in the Caucasian population with a prevalence of 0.3%. They have a reported prevalence of 0.4% in children (30). Seventy percent of drusen are bilateral and may occur as a dominant trait with incomplete penetrance. Associations with retinitis pigmentosa and angioid streaks have been described, but no other associated systemic features are known. They are not associated with neurologic disease, yet may simulate optic disc edema (23,25–27).
It is thought that the formation of ONHD is caused by abnormalities in axoplasmic transport because of a small scleral canal and later axonal degeneration (31). Abnormal metabolism is thought to lead to the deposition of calcium within optic nerve axonal mitochondria. The axons are disrupted and mitochondria are extruded. Further calcium deposition in the extracellular space leads to drusen (32).
Up to 87% of patients with drusen have visual field loss. Visual loss from drusen can be acute or slowly progressive, with nonarteritic anterior ischemic optic neuropathy (NAION) being the most common cause of acute visual loss in these patients (33).
Clinical Evaluation
In affected patients, the configuration of the optic nerve head is variable, and the drusen may be visible on the disc surface or buried within the disc (Fig. 16.5). Differentiation of optic disc edema from ONHD is crucial and can lead to diagnostic uncertainty. Examination techniques commonly performed are optic disc autofluorescence, fluorescein angiography, B-scan ultrasonography (B-USG), and computed tomography (CT) scanning (23,34).
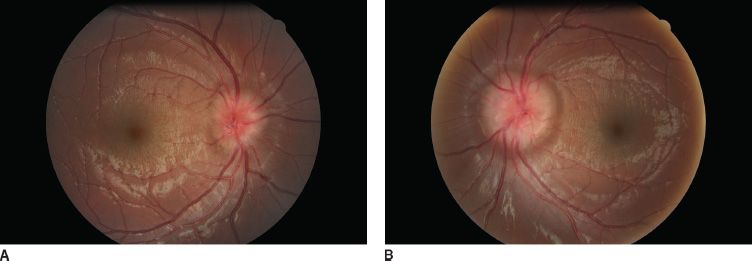
FIGURE 16.5 Bilateral optic disc drusen in a 10-year-old girl. Nasal side of the optic nerve is irregular. Right eye (A), left eye (B).
Mustonen and Nieminen (35) reported that fundus autofluorescence was present in 75% of 180 patients with clinically evident optic disc drusen (Fig. 16.6). In addition, fluorescein uptake with buried drusen can be seen late in angiograms.
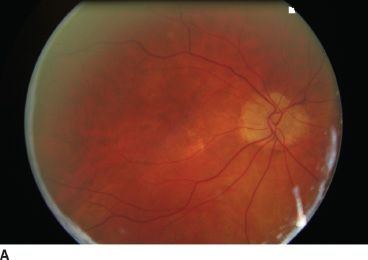
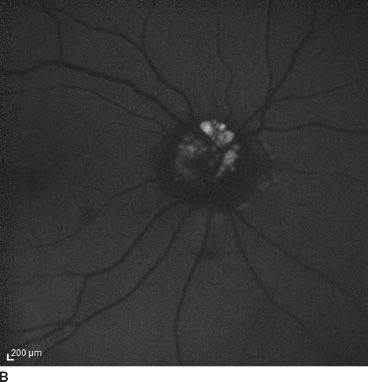
FIGURE 16.6 Color fundus (A) and autofluorescence (B) in the right eye of a 20-year-old patient.
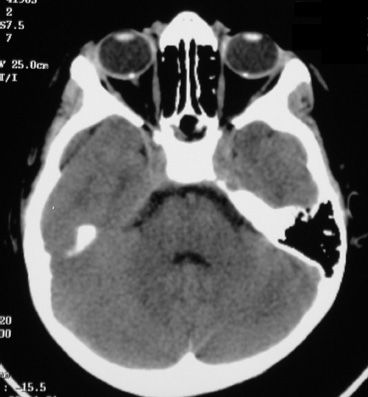
B-USG may detect drusen in 43% of patients with optic disc swelling (36) (Fig. 16.7). Frisen et al. (37) examined five patients with high-resolution CT scan and noted that calcified drusen had increased attenuation within the optic nerve head (Fig. 16.8). Magnetic resonance imaging (MRI) could also be used as a complementary diagnostic instrument showing a surface disc transparency or in order to exclude other optic nerve or brain anomaly. However, it is expensive, requires special techniques and considerable patient cooperation, and is therefore unlikely to be the first choice of investigation in patients with ONHD. B-USG on the other hand is rapid, noninvasive, and inexpensive. Furthermore, B-USG has been shown to be superior to autofluorescence and CT (38).

FIGURE 16.7 Ultrasound of bilateral optic nerve drusen appearing hyperreflective in a 7-year-old boy.
Recently, there have been a number of reports using optical coherence tomography (OCT) to distinguish optic nerve edema from ONHD and that focused on measurements of the peripapillary retinal nerve fiber layer (RNFL) thickness (24,39–42) and direct visualization of the optic nerve head (24).
RNFL thickness, especially in the nasal quadrant, has been shown to be decreased in ONHD when compared with optic nerve edema (24). Lee et al. (24) using SD-OCT detected 80.0% sensitivity and 88.9% specificity for nasal RNFL thickness > 78 mm. Johnson et al. (43) reported 80% specificity and 70% sensitivity for nasal RNFL thickness > 86 mm for the differentiation of ONHD from optic nerve edema (Fig. 16.9). In some patients with ONHD, photoreceptor changes also have been documented (44).
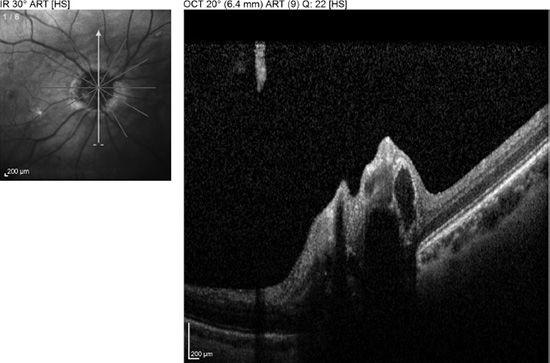
FIGURE 16.9 OCT scan of the head of the optic nerve showing the drusen in the optic nerve and the elevated nerve of patient of Figure 16-6.
Using OCT, Savini et al. (40) identified the subretinal hyporeflective space (SHYPS), a hyporeflective space located between the sensory retina and the retinal pigment epithelium and choriocapillaris in optic disc edema patients. Johnson et al. (43) found a decrease in the mean SHYPS thickness in ONHD patients compared with those with optic nerve edema. Thus, they characterized the OCT appearance of optic nerve edema as an elevated optic nerve head with a smooth internal contour and a SHYPS thickness under the optic nerve head with a gradient taper away from the disc. An irregular surface of the disc creating a “lumpy bumpy” internal optic nerve contour and a more abrupt taper of the SHYPS were suggestive of ONHD (43).
Complications
ONHD have been reported to cause myriad local vascular complications, including central retinal artery and vein occlusion and peripapillary choroidal neovascularization (45).
The presence of bilateral ONHD causing choroidal neovascularization in children is a rare occurrence, with the earliest reported case in a 3-year-old child (46). Peripapillary choroidal neovascularization can cause central vision loss by subfoveal progression of the choroidal neovascularization, serous macular detachment, or submacular hemorrhage (46,47). Various treatment strategies have been advocated. Observation may be a viable option, but there are insufficient data from studies comparing observation with reported treatment modalities.
Laser photocoagulation was reported successful at 2 years by Delyfer et al. (48) as a treatment for two patients with bilateral choroidal neovascular membranes from ONHD and subretinal hemorrhages extending into the macula. Photodynamic therapy with verteporfin (Visudyne; Novartis Ophthalmics, Basel, Switzerland) has also been used. Silva et al. (49) were the first to report successful treatment of bilateral choroidal neovascularization associated with ONHD in a 10-year-old girl. After two sessions of photodynamic therapy, no recurrences were noted after 2 years of follow-up. Surgery is another treatment option. Mateo et al. (50) surgically removed choroidal neovascular membranes in four eyes with ONHD, all of which showed significant visual recovery without any recurrence during 12 to 42 months of follow-up.
Anti-vascular endothelial growth factor (VEGF) agents are now commonly used to treat adults with choroidal neovascularization from a variety of causes (51). Their use in children has been reported in Coats disease (52), Best disease (53), and coloboma-related choroidal neovascularization (54,55). Knape et al. (47) presented a case of bilateral choroidal neovascular membranes associated with ONDH in a 5-year-old boy who was successfully treated with a combination of focal laser photocoagulation and intravitreal bevacizumab. Until the safety of anti-VEGF therapy has been established in the pediatric population, consideration of these medications should be reserved for select cases of choroidal neovascularization threatening central visual acuity.
NAION may occur at an unusually young age in patients with optic disc drusen; it has been described in a 13-year-old child and in several patients in their later teens and early twenties (33,56–58). The visual outcomes in these patients vary, although they are slightly better than for other NAION patients. Some patients do well clinically (56,57). Proposed mechanisms of vision loss include distortion of blood vessels by drusen (33). Systemic hypotension, dehydration, and altitude have been suggested as contributory factors for NAION in the setting of ONDH and may provide an explanation for why patients often discover symptoms upon awakening (59). No effective prevention or treatment for NAION is known. The risk of contralateral involvement is estimated around 15%.
Conclusion
ONHD, despite being a benign condition, have been reported to cause a number of local vascular complications, including central retinal artery and vein occlusion, peripapillary choroidal neovascularization, and NAION. Patients should be aware of these risks and seek ophthalmologic follow-up. We recommend that parents of children with optic disc drusen be instructed about the risk of NAION and potential contributory factors, such as dehydration and altitude, to help prevent vision loss in their children.
Introduction
An optic disc pit is a congenital anomaly of the optic nerve frequently associated with macular detachment (60–62). Pits are usually small, well-demarcated, temporally located depressions in the nerve substance (Fig. 16.10). They may, however, be situated in any sector of the optic nerve head (61). They are sometimes categorized as a subset of atypical optic nerve colobomas (63). Although typically unilateral, bilateral optic pits can be seen in 15% of cases (64).
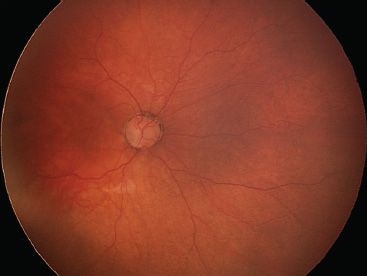
FIGURE 16.10 Optic disc pit in a 4-year-old boy.
Familial cases of optic pits suggest a dominant inheritance pattern (65,66). They have been associated with iris coloboma (67). Optic nerve pits rarely portend additional malformations in the central nervous system (CNS) (68). Acquired pits have been associated with high myopia (69). Their presence among patients with glaucoma constitutes a risk factor for progressive optic disc damage and visual field loss (70) as well as an abnormal susceptibility of the optic nerve to damaging effects of intraocular pressure (IOP) (71). Histopathologic analysis of optic disc pits reveals perineural herniation of poorly differentiated retinal tissue combined with vitreous collagen around the optic nerve into the subarachnoid space (72).
Visual acuity is usually normal. The most common visual field defect is a paracentral arcuate scotoma associated with an enlarged blind spot (61). Acquired visual loss is usually due to the development of maculopathy. Serous macular elevations have been estimated to develop in 25% to 75% of eyes with optic pits (61). The risk is higher in eyes with larger and temporally located pits (Fig. 16.11). When disc pit maculopathy is established, the natural history tends to be poor, with most visual loss occurring in the first 6 months after diagnosis (62). Even if spontaneous resolution of maculopathy does occur in 25% of cases (62), vision often remains poor with the development of outer layer lamellar holes or pigment epitheliopathy. True macular holes are rare (73).
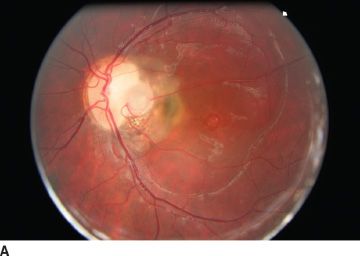
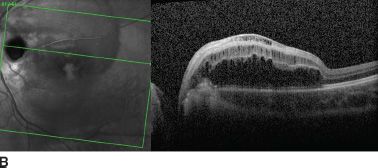
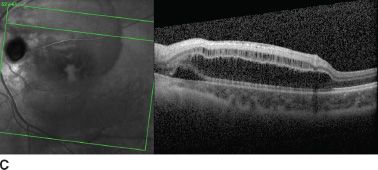
FIGURE 16.11 A: Large optic disc pit in a 10-year-old girl. Persistent maculopathy reduces vision to 20/100. Temporal atrophic and pigmented peripapillary scars of laser photocoagulation. OCT scan shows schisis appearance of retinal layers (B), and localized serous detachment is seen on the superior border (C).
The Optic Pit Maculopathy
The pathogenesis of optic disc pit maculopathy remains unclear. Fluorescence of the area of the disc pit occurs late in fluorescein angiography, and there is no leakage into the submacular fluid. Initially, this was interpreted that the source of macular fluid was not leakage from vessels within the pit itself (74). Cerebrospinal fluid (CSF) pressure is normally lower than intraocular pressure so that flow would normally be expected to occur from the eye to the CSF down a hydrostatic gradient. However, the possibility that CSF could transudate across the fibrous capsule into the retina to cause maculopathy could not be excluded (75). Intracranial pressure and intraocular pressure both fluctuate, although intraocular pressure is usually greater than intracranial pressure. When intraocular pressure is greater, vitreous fluid could move into the pit sac. When intracranial pressure is greater, depending on the body position, for example, some fluid in the sac may be pushed back into the eye through the pit. However, some fluid in the sac also could move into the retina, leading to a retinoschisis-like separation.
Studies in eyes with optic pits using high-resolution OCT pictures identified a membrane crossing the cup of the optic nerve, which may represent neuroectodermal or astroglial tissue (75) (Fig. 16.12). When present, the membrane appears to originate from the edges of the optic disc cup and traverses the cup to join the main vessels at the nasal aspect of the disc or centrally if the vessels emanate centrally from the disc. The thin membrane is concave and roughly follows the contours of the cup. A separation is seen between the membrane and the base of the cup, and fine trabeculae of tissue can sometimes be seen traversing the space, or the space may appear to contain some poorly defined matter.
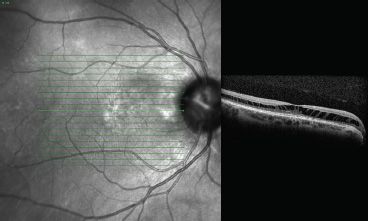
FIGURE 16.12 OCT appearance of maculopathy: extended schisis-like aspect in outer nuclear layers and central serous detachment.
The protective nature of this membrane was suggested by the presence of an intact membrane in patients without maculopathy (75). OCT studies have suggested a communication between the schisis cavity or subretinal space and the disc pit in patients with maculopathy, whereas the retinoschisis-like retinal separation was most prominent in the outer nuclear layers and frequently combined with multiple shallow inner retinal separations (76,77) (Fig. 16.13). It has thus been suggested that a defect in the membrane overlying the disc pit allows fluid to enter the retina causing schisis-like separation of the retinal layers. True macular detachment can then occur as a secondary event but can also occur in the absence of associated retinal thickening and fluid within the retinal layers (76).
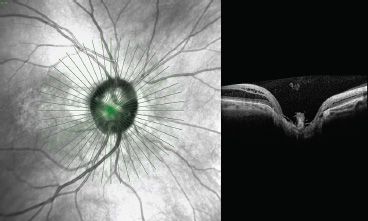
FIGURE 16.13 OCT scan of the optic nerve of Figure 16-12 patient showing prepapillary glial material.
A role for vitreous traction has also been postulated from the observation that maculopathy becomes asymptomatic in the fourth decade of life and that it can resolve after spontaneous posterior vitreous detachment (PVD) (78). Bonnet (78) previously reported that none of the eyes with macular detachment in their series had evidence of a PVD and that the two eyes that demonstrated spontaneous reattachment did so after a PVD developed. In addition, Theodossiadis et al. (79) described vitreous abnormalities such as vitreomacular traction and vitreous strands over the optic disc associated with optic disc pit maculopathy. Hirakata et al. (80) in their study equally proved that vitreoretinal traction is an important factor in the pathogenesis of optic disc pit maculopathy and that induction of PVD relieving any anteroposterior traction on the macula is effective in reversing macular elevation without the need for laser treatment. During induction of a PVD, a tight adhesion of the posterior hyaloid or abnormal membrane, such as anomalous Cloquet canal, to the margin of the disc pit has been reported (81,82).
When there is posterior hyaloid traction caused by age-related vitreous liquefaction or trauma, it pulls on the optic disc pit. Because the pit is pulled up like a tent by vitreous traction, the turbulent fow may have increased access around the vessels to the intraretinal space. This traction affects the peripapillary retina, especially around the pit. This further facilitates intraretinal fluid accumulation. When a PVD occurs, the anterior traction is released. The pit decompresses and moves downward, and access to the subretinal space becomes limited. Any traction on the peripapillary retina also is released.
Treatment
Patients normally seek ophthalmologic evaluation because of a diminished visual acuity or the discovery may be made incidentally during a routine ophthalmologic assessment. When an optic pit is detected, performing an OCT can be very useful in order to depict the presence of a PVD, a schisis-like separation, and/or subretinal fluid. In cases with severe maculopathy associated with vitreomacular traction, vitrectomy allows release of the vitreoretinal traction and should be routinely performed. The use of a gas tamponade agent may not be necessary for success (80). In fact, subretinal migration of gas or silicone oil has been reported (83,84). Theoretical calculations suggest that the pressure differential required for migration of gas through a small defect in the roof of a cavitary disc lesion is within the range of expected fluctuations in CSF pressure. Explanations should be given concerning the nature of this pathology and the visual acuity outcome.
Conclusion
Optic disc pits are usually unilateral, sporadic, and not associated with systemic disease. The etiology of the associated serous maculopathy still remains unclear. It could be suggested that the unusual movement of fluid between the vitreous cavity and subarachnoid space and the traction around the posterior hyaloid may contribute to the pathogenesis of this disease. Vitrectomy with induction of a PVD without a gas tamponade or laser photocoagulation allows resolution of optic disc pit maculopathy in most cases.
Introduction
Colobomas of the optic nerve result from incomplete or abnormal closure of the primitive embryonic fissure (85). In certain instances, the defect may extend to involve the choroid, retina, ciliary body, and iris. The term “coloboma” derives from Greek and means “defect” (from the adjective “colobos,” which means “mutilated” or “cut short”).
Optic disc coloboma comprises a clearly demarcated bowl-shaped excavation of the optic disc, which is typically decentered and deeper inferiorly (86). The coloboma occupies the lower part of the optic nerve head. The neuroretinal rim is absent inferiorly but is usually identifiable superiorly (Fig. 16.14). In cases in which the adjacent inferior retina and choroid are deficient, microphthalmia may also be evident (87). Optic disc colobomas may appear as large excavations of the nerve head up to 25 diopters deep and involving nearly all of the disc, which may be enlarged (88).
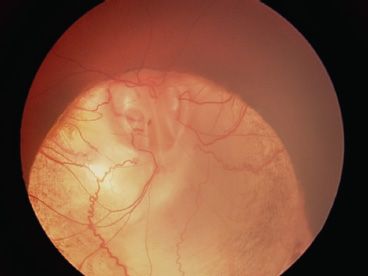
FIGURE 16.14 Optic nerve coloboma in a 16-year-old patient having lost her left eye by retinal detachment. Macular pigment is visible on the edge of the coloboma, and vision is 20/100.
Optic nerve colobomas are usually unilateral; bilateral colobomas are frequent but asymmetrical (88). Significant refractive error and anisometropia are common. The only feature that relates to visual outcome is the degree of foveal involvement by the coloboma (89). The size of the coloboma, the color of the optic nerve, and the presence of subfoveal pigment change are not related to visual outcome (Fig. 16.15).
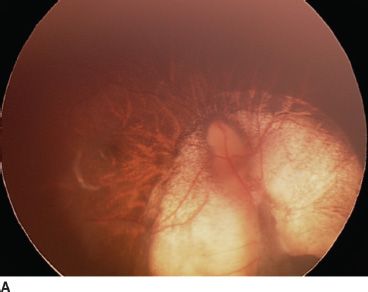
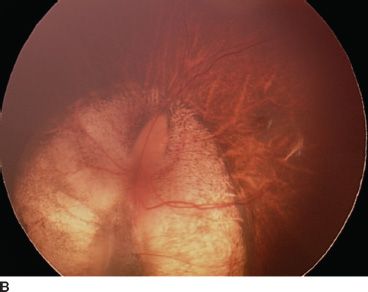
FIGURE 16.15 Bilateral optic nerve coloboma sparing the macula in both eyes. Vision is 20/20 in this 18-year-old patient. Right eye (A), left eye (B).
Most colobomas are sporadic, although an autosomal dominant hereditary pattern has been reported and less frequently an autosomal recessive or X-linked recessive pattern (88,90). The condition can occur in association with multiple congenital abnormalities indicative of an insult to the developing fetus during the 6th week of gestation (Fig. 16.16).
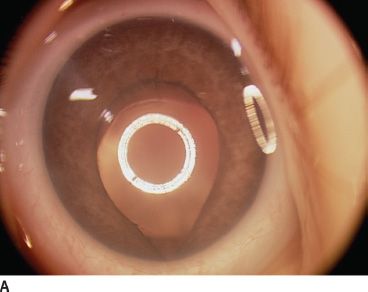
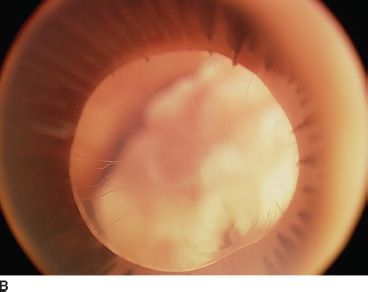
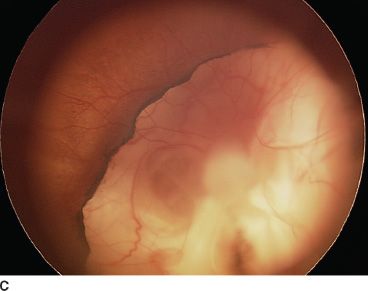
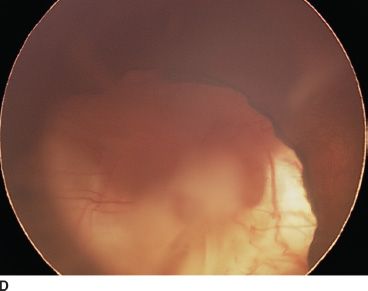
FIGURE 16.16 An 18-month-old child presenting with bilateral microphthalmia and coloboma. A: Immature iris and inferior coloboma. B,C: Microspherophakia with extended ciliary processes and posterior persistent fetal vascularization (fibrous stalk) attached to the large chorioretinal coloboma. D: Chorioretinal coloboma of the fellow eye.
There is a wide range of associations. These have been reviewed by Brodsky (87) and include the CHARGE association (coloboma, choanal atresia, congenital heart disease, and multiple other abnormalities), Walker-Warburg syndrome, Goltz focal dermal hypoplasia, Aicardi syndrome, Goldenhar syndrome, and linear sebaceous nevus syndrome. More recently, associations with Dandy-Walker malformation (91) and renal coloboma syndrome (with a mutation of PAX2 transcription) (92–95) have also been described. In the latter, the renal disease may vary significantly from patient to patient. Associated abnormalities include small dysplastic kidneys, vesicoureteral reflux, and high-frequency hearing loss. While the renal disease may be quite variable, all patients identified with mutations in the Pax2 gene have been observed to have colobomatous optic nerves. Iris colobomas have not been documented with Pax2 mutations.
Expression of PAX2 is restricted to cells of astrocytic lineage both during retinal development and in adulthood. Using immunohistochemistry, it has been found that adult retinal cells with the antigenic phenotype present in mature perinatal astrocytes are found only in the region surrounding the optic nerve head and that astrocyte precursor cells expressing PAX2 are found in a small region surrounding the optic nerve during early development. It has been argued that these findings suggest that coloboma formation may be associated with impaired astrocyte differentiation during development (96).
The association of optic nerve coloboma with dysgenesis of the internal carotid artery and transsphenoidal encephalocele with hypopituitarism has led to the suggestion that the link between these malformations is abnormal neural crest cell development (97).
Recently, more details came to light concerning the anatomical aspect of the coloboma; high-resolution OCT scanning has detected a membrane overlying the disc coloboma of some patients without maculopathy, which probably represents the neuroectodermal lining of the coloboma (75). Discontinuities of Bruch membrane and retinal pigment epithelium at the margin may allow subpigment epithelial and choroidal vessels to enter the subretinal space at this junction. T1 MRI confirms that the intracranial portion of the optic nerve is reduced in size (98)
Coloboma-Related Complications
The presence of an optic nerve coloboma implies some possible complications. A small proportion of cases are associated with cysts arising from the optic nerve sheath, which communicate with the subarachnoid space (99). Rarely such cysts can enlarge and lead to compressive optic neuropathy (100). Peripapillary choroidal neovascularization has been described in association with optic nerve coloboma (101,102). Studies have shown good response of this type of neovascular membrane to treatment with intravitreal bevacizumab (53).
Retinal detachment occurs in approximately 45% of patients with optic nerve coloboma (62) (Fig. 16.17). The source of the subretinal fluid is not known but could derive from fluid entering the retrobulbar space from surrounding orbital tissue, or from the choriocapillaris, or from CSF. In contrast to retinochoroidal colobomas, rhegmatogenous detachment is probably not a recognized association (103). The retinal detachments associated with optic nerve colobomas typically are more extensive and more bullous than those caused by optic nerve pits, which affect only the macular area and are often accompanied by schisis. The pathophysiology appears to be similar in both of these conditions, with a defect in the abnormal papillary tissue allowing fluid to communicate to the subretinal space and detach the retina. In patients with an optic nerve head coloboma, the extent of abnormal tissue is much greater than it is in optic pits. This papillary defect along with a poorly differentiated retina and retinal pigment epithelium, an abnormal lamina cribrosa, and an enlarged scleral canal potentially allows the vitreous cavity, subretinal space, subarachnoid space, and orbital space to communicate in these patients (104). If a defect develops and liquefied vitreous has access to the hole, then the retina detaches. Circumferential intrascleral smooth muscle has been observed histologically (105) and may account for the rare observation of spontaneous contractility of the colobomatous optic disc (106). Spontaneous reattachment, however, may occur (107).
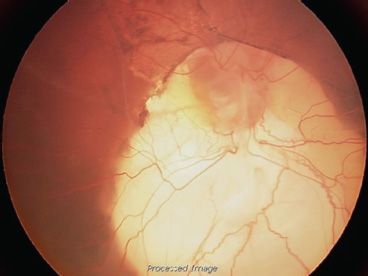
FIGURE 16.17 Retinal detachment complicating a large optic nerve coloboma in a 6-month-old boy. The presence of folds involving the macula and pigment epithelial changes favor the hypothesis that this was a congenital retinal detachment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


