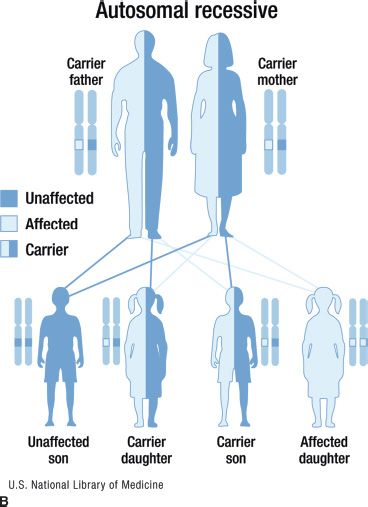
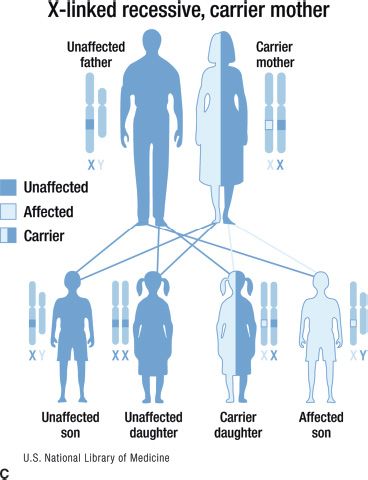
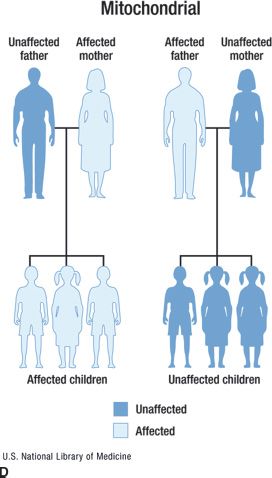
FIGURE 15.1 Inheritance patterns for genetic conditions with Mendelian inheritance. This image is a work of the National Institutes of Health, part of the United States Department of Health and Human Services. As a work of the US federal government, the image is in the public domain.
Autosomal recessive disorders are those in which an individual must have mutations in both copies of the disease gene in order to be affected. The individual may be homozygous (two copies of the same mutation) or compound heterozygous (a different mutation in each copy of the disease gene). Carriers have only one copy of the mutation and are usually asymptomatic with a normal phenotype. Both parents of a child affected with an autosomal recessive condition are generally carriers, and each of their offspring has a 25% chance of having the disorder. Multiple family members in a single generation (e.g., siblings) may be affected, but vertical transmission is not commonly seen in autosomal recessive conditions as shown in Figure 15.1B. All children born to a parent affected with an autosomal recessive condition will at least be carriers of the condition. Their children may be affected if the other parent is a carrier, which is generally unlikely given the rarity of these conditions, but the probability is increased if the parents are related or from an ethnic background with a relatively high prevalence of the disorder. Stargardt disease and Usher syndrome are conditions that have autosomal recessive inheritance patterns. Stargardt disease, caused by ABCA4 mutations, is a juvenile macular dystrophy and has a relatively high carrier frequency. The carrier frequency of ABCA4 mutations in the general population is approximately 1/50.
An X-linked disorder is one in which the genetic mutation is on the X chromosome. Because males have only one X chromosome, all males with the mutation are affected with the disorder. Female have two X chromosomes; thus, if they have one copy of the mutation, they are carriers. For an X-linked recessive disorder, female carriers are usually not affected, although they may display some symptoms due to skewed X inactivation. Generally, these symptoms are milder than those seen in affected males. Fathers do not pass an X chromosome to their sons, so the lack of male-to-male transmission is characteristic of X-linked disorders. All daughters of a man with an X-linked recessive condition will be carriers. When the mother is a carrier, the risks to her children depend on their gender: sons of carrier females have a 50% risk of being affected, whereas daughters of carriers have a 50% chance of being carriers as shown in Figure 15.1C. Juvenile retinoschisis caused by mutations in the RS1 gene is a condition that follows an X-linked recessive inheritance pattern. Female carriers of an RS1 mutation rarely express fundus abnormalities although clinical findings have been reported in a few cases (3). While rare, some conditions display X-linked dominant inheritance in which females with one copy of the mutation are affected. X-linked dominant conditions are often embryonically lethal in males. An important example of an X-linked dominant condition is incontinentia pigmenti, which has both skin and ocular manifestations.
Genetic conditions can also result from mutations in mitochondrial DNA (4). Mitochondria are exclusively inherited from the mother, and family histories with a pattern demonstrating mitochondrial inheritance will reveal an affected mother with affected children of both genders, as shown in Figure 15.1D. The presence of affected females as well as affected males can help distinguish this inheritance pattern from X-linked inheritance. Complicating mitochondrial inheritance is the presence of heteroplasmy, which occurs when cells have some normal and some mutated copies of their mitochondrial DNA. In general, an individual will only show symptoms of a mitochondrial condition if the proportion of mutated mitochondrial DNA reaches a certain threshold. Therefore, the disease could be clinically absent in a woman, but present in both her mother and her children. Mutations in mitochondrial DNA can cause ocular diseases such as Kearns-Sayre (associated with retinal degeneration) or Leber hereditary optic neuropathy (LHON). A male predomination in LHON has been observed, but the cause is currently unknown.
A disease is described as sporadic if the affected individual has no other family members with the disorder. Some sporadic conditions may be the result of mutations in a single gene, and others may have environmental causes. If the clinical findings are consistent with a known Mendelian disorder, then the sporadic appearance is often due to a germline de novo mutation; thus, individuals with a sporadically appearing genetic disorder can be at risk of passing the disorder to their offspring. An example of a condition that may be sporadic is aniridia, which is associated with mutations in the PAX6 gene. Each offspring of an individual with a de novo PAX6 mutation has a 50% chance of inheriting the mutation and being affected with the disorder. In contrast, the individual’s parents and siblings have minimal risk of having an affected child. Note that for an autosomal recessive disorder, there is often only one affected member of the family; thus, the condition may appear to be sporadic.
Many conditions are influenced by environmental factors as well as genetic mutations, such that risk of disease is increased with exposure to the environment. A trait or condition is said to have a multifactorial inheritance pattern if both genetic and environmental factors contribute to the phenotype. Traits such as blood pressure and height are common examples of multifactorial inheritance. Many ophthalmic conditions are multifactorial. For example, age-related macular degeneration (AMD) has both genetic and environmental factors that contribute to disease development, progression, and severity. Recognized environmental risk factors for AMD include age, hypertension, smoking, diet, obesity, and chronic inflammation (5). Genetic contributions to AMD have been demonstrated in both family and twin studies (reviewed by Chamberlain et al. [6]), and the multifactorial nature of the condition was highlighted. There are known genetic variants that predispose a patient to developing AMD and can be identified through genetic testing. As our knowledge of ophthalmic conditions expands, both the genetic and the environmental factors that contribute to the phenotypes will be better understood.
Determining the pattern of inheritance can be complicated by incomplete penetrance and variable expressivity. Penetrance is a statistical term; it is defined as the probability that a person with the abnormal genotype expresses the phenotype, or the proportion of individuals in the population with the disease-causing mutation, who also have the clinical symptoms of the disease. Penetrance is usually complete in recessive conditions but can be <100% in autosomal dominant disorders, meaning that some individuals who have the abnormal genotype are not affected. For genetic disorders with adult onset, penetrance is age-dependent. Forms of autosomal dominant retinitis pigmentosa with incomplete penetrance have been reported. Variable expressivity, on the other hand, is a descriptive term for the variation in phenotype among individuals with the abnormal genotype (7). There is usually greater phenotypic variation in autosomal dominant conditions than in autosomal recessive or X-linked disorders.
Family History
The family history is an important tool for determining differential diagnoses, risk assessment, and possibly medical management recommendations. Additionally, the family history allows the genetic counselor to assess patient education needs and psychosocial influences. A family history is obtained in virtually all genetic counseling sessions, generally includes at least three generations, and often assesses a variety of health concerns. For this reason, the family history is arguably one of the most important tools in both screening and prevention of common diseases (8,9). Family histories begin with a consultand (the individual seeking genetic consultation) and then include first-degree relatives (parents, siblings, children), second-degree relatives (aunts/uncles, nieces/ nephews, grandparents/children), and often third-degree relatives (cousins, great aunts/uncles). When relevant health information about more distant family members is known, this information should be included in the family history as well. Targeted questions regarding the status of affected as well as unaffected individuals can help the genetic counselor determine the most likely diagnosis as well as risk to other family members. Inheritance patterns and disease-specific risk assessments can be generated from family history alone in many cases.
The family history and/or pedigree should be an integral part of the medical record. The term pedigree refers to the way the family history is visualized. Pedigrees should be drawn in a standard fashion to allow for easy interpretation by other health care providers. Guidelines for pedigree documentation have been developed by the National Society of Genetic Counselors (10). Figure 15.2 provides an overview of the most commonly used pedigree symbols. Full names, dates of birth, or any other potentially identifying information should be excluded from the pedigree in order to maintain the confidentiality of family members. How to best capture family history information in the electronic medical record (EMR) is the subject of a great deal of research. For the past several years a variety of institutions and researchers have worked to create a pedigree tool that is compatible with the EMR; however, a widely agreed upon tool does not yet exist.
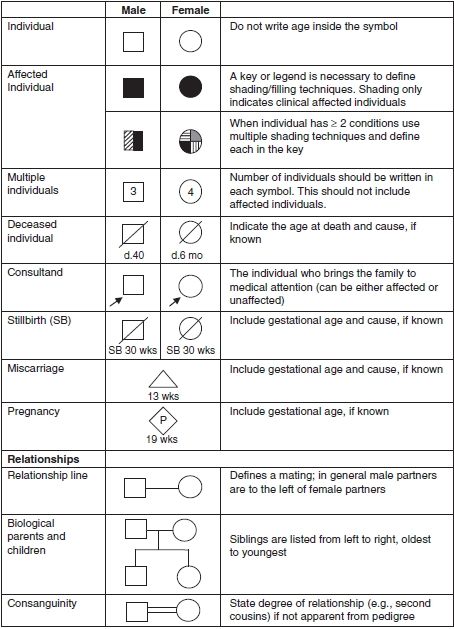
FIGURE 15.2 Common symbols used in pedigree construction. (Adapted from Bennett RL, French KS, Resta RG, et al. Standardized human pedigree nomenclature: update and assessment of the recommendations of the National Society of Genetic Counselors. J Genet Couns 2008;17:424–433.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


