Endothelium Growth Factor
Pharmacological Agents
Leanne T. Labriola • Muge Kesen • Karl G. Csaky
Introduction
Revascularization is a serious sequela of ocular diseases including age-related macular degeneration, diabetic retinopathy, vascular occlusive disorders, and retinopathy of prematurity. Research has identified vascular endothelium growth factor (VEGF) as one of the main signaling proteins in this process. With this discovery, novel strategies of preventing neovascularization have focused on inhibiting the VEGF molecule. This has led to a new pharmaceutical class of anti-VEGF drugs, including pegaptanib, ranibizumab, and bevacizumab. This chapter discusses the pharmacokinetic and pharmacodynamic properties of these drugs.
Clinical Pharmacology—Background
Pharmacokinetics and Pharmacodynamics
Pharmacokinetics interprets how the different tissues and complexes within the body chemically alter the drug. It studies the drug path from its entry to its excretion from the body. Pharmacokinetics includes the mechanisms of drug liberation, absorption, dispersion, metabolism, and elimination from the body. Alternatively, pharmacodynamics is the study of the cascade of reactions from the drug’s initial activation as a drug-receptor complex to its final downstream biological effect. Pharmacodynamics identifies how drugs stimulate, depress, destroy, or replace other substances or reactions in the body.
Compartment Models
One goal of pharmacokinetics is to define the drug concentration as a unit of time. Two different rationales are used to accomplish this goal. They include the noncompartmental model and the compartmental model. These models use different equations to estimate the amount of available drug in the body. The validity of the noncompartment model relies on the estimation of the entire dose of the drug. It uses this amount to create an area under the curve (AUC) graph of the concentration verses time. Direct measurements of drug concentration at several time points increase the accuracy of the curve.
The two-compartmental model uses kinetic theory to estimate the concentration of the drug, which is then entered into a linear or exponential equation to plot the concentration verses time graph. Different equations are used to estimate the total amount of drug as it enters and distributes through the central compartment as compared to when it redistributes to the peripheral compartments. Since the distribution of the drug into the various compartments takes place at the same time as the drug is being eliminated from the body, these models are highly complex.
To use these models in research studies, certain assumptions are made. The first is to assume that the magnitude of the effect is proportional to the amount of drug present. The second is to assume that the drug creates a drug-receptor complex that is equal to the total effect of the drug. The third is to assume that the total amount of the receptor is constant. And finally, the fourth is to assume that the concentration of drug in the plasma is equal to the amount of unbound drug in the body. For the study of the pharmacology of the anti-VEGF agents, most of these assumptions have yet to be verified.
Ocular Pharmacology
Models for Studying Ocular Pharmacology
Initial studies on the pharmacokinetics of intravitreal drugs have been established using animal models. Animal models can provide basic concepts of drug functioning and identify overall routes of metabolism, distribution and eliminations. In this chapter, we report on pertinent findings from animal studies and explain their contribution to our current understanding of anti-VEGF medications. However, the use of animal models has several limitations when used as a comparison model for human tissue. These include the differences in anatomical size, tissue properties, and structure. In addition, animal tissue can be damaged or altered from the methods used to harvest and collect the tissue and the need to euthanize the animals to collect data over several time intervals weighs against the overall utility of this method of research.
The size of the eye and its components vary among different species. In current literature, aqueous volume in the rabbit eye is estimated to be 0.251 and 0.134 mL in monkeys.2 These values are similar to human aqueous chamber of 0.25 mL. However, in humans, the vitreous chamber is 4.5 mL. While rats have a vitreous volume of 56 μL, rabbits and monkeys have a vitreous volume ranging from 1.5 to 3 mL, depending on the species (Fig. 13.1).3
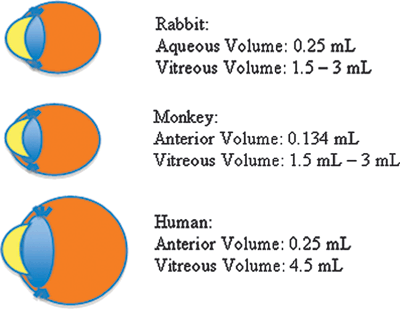
FIGURE 13.1. Aqueous and vitreous volumes of rabbits, monkeys, and humans.
Another disadvantage is that the components of the vitreous can be different between species, which creates different inherent viscosities and other tissue properties 4 leading to different volumes of distribution in humans, monkey, rabbits, and rats. The structural relationships between tissues are also different between species. For example, the rabbit eye has no fovea.5 In regard to the vascular system of the retina, the rabbit retina is merangiotic, or partially vascularized, while human retinas are holangiotic, or completely vascularized.6 Humans have uneven distribution of blood supply to the retina, with higher vascular flow to the macula and lower flow to the peripheral retina. Humans also have higher choroidal blood flow rates than animals. These differences in blood flow would affect the speed and extent of intravascular delivery or elimination of substances and provide a confounding variable when comparing blood flow between species.
An advantage of animal models is that the tissue can be harvested at different time points and analyzed in various preparations. However, this process also has its limitations. For example, with tissue preparation, the dynamic relationships between the tissues are lost, the three-dimensional structures are changed, and the tissue becomes a static representation of one segment of time within the study. These changes due to the tissue collection influence the interpretation of the tissue and limit the utility of the final results.
In conclusion, although animal models are advantageous in the fact that they can offer controlled experimental conditions and more invasive strategies can be used for measuring the amount of drug within the tissues, caution has to be used when extrapolating information gained from the studies.
New Technology
New noninvasive modalities are being developed for imaging proteins in the vitreous through radioactive labeling. One example is dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI). This technology uses MRI scans to acquire three-dimensional images in vivo in order to better understand spatial relationships and flow of substances. DCE-MRI is being used in eye research to delineate episcleral outflow pathways, which is just beginning to elucidate the value of this imaging modality in research studies.7 Similar to DCE-MRI, positron emission tomography uses stereotactic fiduciary markers; specifically,18 F-fluoromisonidazole and single photon emission computer tomography (SPECT) offer dynamic images of the microenvironment of tumor cells.8 These images are able to identify the hypoxic centers of tumor cells and may be used in ocular images in the future.
Anti-Vegf Drug Pharmacology
Ocular Barriers to Anti-VEGF Drug Distribution
A major barrier to the flow of substances within the eye is the existence of multiple basement membranes, which include the internal limiting membrane (ILM), the external limiting membrane (ELM), and Bruch membrane. Both the extent of flow impediment and the mechanism in which these membranes can limit flow are still unknown. Contemporary research studies offer opposing theories on these issues. Some authors believe that the membranes can significantly, if not completely, stop molecules from penetrating through the layers of the retina, based on the molecular size. Other groups have shown with their research that even large molecules, such as antibodies, can completely pass through the retina from the inner to outer layers, even going into the choriocapillaris.
The ILM is made from the footplates of Müller cells and attachments to the basal lamina. This structure separates the vitreous from the nerve fiber layer of the retina. Debate exists as to the amount and type of restriction that is created by the ILM. Some research findings suggest that the ILM acts as an impenetrable mechanical filtration barrier as well as a formidable electrostatic barrier,3,4,9,10 while others studies have show that even large molecules are able to effectively penetrate the ILM.11 The existence of pores ranging from 10 to 25 μm in size within the ILM9 supports the argument of a mechanical barrier for this layer. Ranibizuamb, the Fab fragment of bevacizumab, an antibody to VEGF, has been shown to penetrate all layers of the retina, while, a full-sized antibody against the human epidermal growth factor receptor 2 (HER2) could not pass through the layers of the retina.3 However, critics of the study state that the two groups of comparison were not equal since two different antibodies, the anti-VEGF and anti-HER2, were used. The HER2 is present in many structures within the retina and specific antibody-receptor binding could have occurred at the boundary of the inner retina and limited the overall penetration of the substance. Another study using intravitreal injections showed that the ILM restricted the passage of a 70-kDa molecule of tissue plasminogen activator (tPA), whereas the 20-kDa rhodamine B molecule was able to penetrate this barrier.10 However, specific binding of one substance and not the other could also have influenced these results since different molecules were compared.
Contrary to the suggestion of the ILM as a mechanical barrier, studies have shown that large molecules, including high molecular weight antibodies, are in fact able to pass through all of the layers of the retina. These molecules include albumin,12 a tPA analogue called tenecteplase,13 antibodies against recoverin,14 melanoma-associated retinopathy antibody,15 and others.5,11
On the outer side of the retina, the zonular adherents between the adjacent photoreceptors and Müller cells form the ELM. This is not a true histological membrane and many fenestrations exist within it; however, some authors suggest that this membrane can also offer mechanical or electrostatic resistance to substance flow within the retina.9
Deep to the retina, Bruch membrane provides yet another barrier to unwanted substances. It is located between the retinal pigment epithelial cells and the choroid. Bruch membrane specifically has been reported to limit the diffusion of macromolecules, including proteins, oglionucleotides, and genes, from entering the retina.3,9,16 However, more research needs to be done to delineate the path of flow of molecules that can penetrate the retina and investigate if they disrupt Bruch membrane or use transport mechanisms to cross through it.
Within the eye, the vitreous provides its own type of impediment to the free flow of substances. The vitreous occupies four fifths of the volume of the globe. The vitreal collagen network of fibers and proteoglycan elements creates a barrier to the diffusion of molecules from the anterior hyaloid face to the posterior vitreoretinal surface. The composition of elements include hyaluronan, heparin sulfate, dermatan sulfate, and chondroitin sulfate, which convey an overall negative charge to this matrix, which like the ILM and ELM can create not only a mechanical barrier but also an electrostatic one as well.9
Anti-VEGF Ocular Drug Delivery
To date, all of the anti-VEGF agents are delivered by direct intravitreal injection. The absorption of drugs delivered to the eye is influenced by the drug’s molecular mass, molecular radius, and solubility. Intravitreal injections essentially have 100% bioavailability to ocular tissues since they are injected at their site of action.
Once the drug is inside the eye, it begins to distribute throughout the vitreous. Fluorescein labeling of molecules and DCE-MRI imaging have shown that molecules exhibit an anterior to posterior concentration gradient within the vitreous humor from a high concentration right behind the lens to a low concentration near the vitreoretinal interface.7,9 As mentioned earlier, the ocular barriers limit drug dispersion within the eye. The properties of the drug, such as the size, the molecular charge, and the diffusion co-efficient, collectively determine the drug’s ability to penetrate the ocular barriers. Recent studies have shown the ability of even larger molecules to pass through the retinal layer, including the blood-ocular barrier. Galbumin, a bovine albumin molecule conjugated with gadolinium, has a molecular weight of 80 kDa and still is capable of penetrating all the layers of the retina. Immunohistochemical tracers and radioactive labeling have been used to confirm the complete transretinal penetration of bevacizumab.11,17,18 Although the mechanism of retinal penetration has not yet been clearly defined, there is evidence to support an active transport mechanism, similar to that of the nanoparticles, such as endocytosis or transcytosis.9,17 It is important to understand how larger molecules, such as antibodies, distribute throughout the vitreous since current therapeutic strategies utilize these molecules to deliver target-specific pharmacotherapy.
Drugs that are not metabolized can be eliminated from the eye. Most studies support a first-order elimination model for ocular clearance. However, others have shown that the elimination of different doses of macromolecules, specifically Galbumin, occurred at a constant rate. The terminal vitreous elimination half-lives of the 1 and 0.25 mg/mL of Galbumin were 2 and 0.7 days, respectively.4 The elimination process showed a steady elimination rate; therefore, this molecule follows a zero-order elimination rate, which is similar to how alcohol is eliminated from systemic circulation.
The major pathways of intraocular drug clearance take place through an anterior route and a posterior route within the eye. The anterior route occurs by the passive diffusion of the drug around the lens into the aqueous chamber and then its excretion via Schlemm canal or the uveoscleral outflow pathway.19–21 Hydrophilic substances favor this anterior elimination route.4 The rate of elimination via this route is dependent on the molecular weight and the solubility of the substance. Clearance through the trabecular meshwork and the uveoscleral outflow depends on the permeability of the trabecular meshwork and the episcleral venous pressure. This route can be affected by the factors that alter both the trabecular meshwork or anatomy of the angle of the anterior chamber and the episcleral venous pressure. For example, the rate of clearance can change with inflammation or the disruption of the blood-aqueous barrier in the ciliary epithelium.22 Overall, this pathway can be slower than the posterior elimination pathway.22 In spite of this, the anterior elimination route is the major elimination pathway of substances clearance from the vitreous.19,21
Medications can also exit the eye through posterior elimination pathways. Posterior pathways include transretinal migration as well as receptor-mediated transcytosis.7,23 Lipophilic molecules can be more easily excreted through this route since they have to pass through the hydrophobic retinal layers.4 Drugs that exit through the posterior route do not diffuse directly through the sclera. Instead these substances are eliminated by outflow to through the choroidal circulation.7 The dynamic properties of the choroid including hydrostatic forces, oncotic pressures, and the fast rate of blood flow combined to eliminate substances through the vascular network of the choriocapillaris. The higher the hydrostatic pressure within the vitreous, the greater the outflow to the episcleral space. In addition, the high extracellular protein levels in the choroid elevate oncotic pressures outside of the vitreous and favor fluid flow across the retina and into the choroid. Lastly, the rapid movement of choroidal blood flow quickly removes the drug from the orbit and expels it through systemic circulation.7 More research needs to be done to elucidate the exact mechanism of this posterior transport and to delineate the method in which substances can evade the barriers of the posterior blood-ocular barrier.
A specific route for posterior elimination has been shown to be through receptor-mediated transcytosis by the Fc receptor found on tissues that constitute the blood-ocular barrier, such as the surface of the retinal epithelial cells. The Fc Receptor can sequester the drug or help to excrete the drug from the eye. The receptor sequesters the IgG antibody within the endothelium of vessels. This sequestering would extend the half-life of the antibody by preventing its catabolization.17 In addition, the Fc receptor can aid in transporting the drug through the ocular barriers to eliminate it from the eye. The Fc receptor is able to perform this role in other tissues outside of the eye. In the brain, the Fc receptor can transport IgG molecules by transcytosis across the blood-brain barrier and deliver the entire antibody intact into the bloodstream. In the eye, the Fc receptor was found on lymph vessels and on the epithelial lining of the blood-ocular barrier, a finding that correlates with the presumed role of the receptor of assisting in elimination of drugs through these barriers.7,23 The discovery of Fc receptors may open a launching platform for new research in ocular pharmacokinetics, which may lead to the identification of other receptor-based transport mechanisms. Research in this field may help to clarify questions on if free drug passage through the blood-retinal barrier is even possible or if all posterior elimination of drugs is receptor-mediated.
Pharmcaokinetics of Current Anti-Vegf Medications
Pegaptanib
Pharmacokinetics
Pegaptanib sodium (Macugen, OIS-Eyetech, NY, USA) is a 28-ribonucleotide molecule with an osmolarity of 280 to 360 mOsm/kg and pH of 6.0 to 7.0 (Drug Insert 2009). Pegaptanib is an aptamer, which is a highly selective derivative of a nucleic acid. Aptamers function by folding into a three-dimensional structure that binds to and directly inhibits the target protein.24 The target site for pegaptanib is the heparin-binding domain of the proinflammatory VEGF165 slice isoform. This region is responsible for intensification of the interaction between the VEGF protein and its receptor that promotes neovascularization. Similar to other aptamers, pegaptanib has a high dissociation constant of 50 pmol/L, making it very effective in binding to VEGF165 and inhibiting the amplification of the VEGF receptor signaling cascade.
The high affinity of pegaptanib for its target isoform limits its effects on other splice isoforms of VEGF, such as VEGF120, which may create a stable balance of angiogenic precursors and contribute to the overall safety of the drug. In addition, because pegaptanib is an RNA-based molecule, it has little to no toxicity or immunogenicity making it an advantageous therapeutic choice (Fig. 13.2).
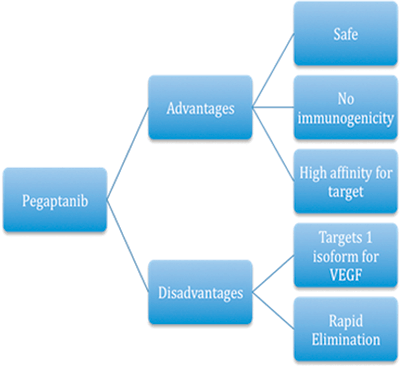
FIGURE 13.2. Advantages and disadvantages of pegaptanib.
Aptamers, however, are limited in their utility by the small molecular sizes, typically 10 to 20 kDa.24 Their small size allows for rapid elimination from circulation. To address this problem, pegaptanib, has been chemically conjugated to two 20-kDa monomethoxy-polyethylene glycol molecules increasing its overall size and molecular weight to 50-kDa. The addition of these molecules to a compound, known as pegylation, limits glomerular excretion and increases the circulatory half-life of the compound.24 Furthermore, pegylation is thought to increase the stability of pegaptanib and alter its distribution to preferentially target highly vascularized organs.24
Absorption
Pegaptanib has a slow systemic absorption, which is the rate-limiting step for this drug.25
Distribution
Initial studies in Rhesus Monkeys using radioactive markers provided early information on the pharmacokinetics of the drugs. After injection of 1 and 2 mg of pegaptanib, the vitreous concentrations were 26 and 499 mg/mL, respectively. The vitreous terminal half-life was 94.1 hours for the 1-mg dose and 89.9 hours for the 2-mg dose.26 This finding reflects a dose-dependant effect of the terminal vitreous half-life of the drug. Twenty-four hours after its administration intravitreously, the drug was detected in the retina, vitreous humor, and aqueous humor within the eye. Twenty-four hours after intravenous administration, the highest concentration outside the eye was found in the kidney.26 In a rabbit model, an intravitreal injection of 0.5 mg of pegaptanib resulted in initial vitreous of 350 mg/mL, which decreased to 1.7 μg/mL by day 28 of the study.26 Pegaptanib showed first-order kinetics and was completely eliminated by 84 hours.26
After the VISION trial confirmed the safety of the drug, studies were conducted in human patients. A single monocular dose of 3 mg of pegaptanib sodium (ten times the recommended FDA dose) was injected intravitreously. Even at this high dose of pegaptanib, the maximum plasma concentration was only 80 ng/mL, which was reached 1 to 4 days after injection of the drug. Using this information to create an AUC graph and extrapolate the information for other doses of pegaptanib, it was found that an intravitreal injection of 0.5 mg would result in a maximum plasma concentration <10 ng/mL. The plasma half-life of the drug was 10 ± 4 days, and there was no accumulation of the drug in the plasma even after six consecutive weekly doses.25,26
Elimination
Because pegaptanib is made from nucleic acids and their site of action is extracellular, it is easily metabolized by the action of endonucleases and exonucleases.25,26 The accessibility of this drug to degradation processes may explain the reason why pegaptanib does not accumulate in the serum. Pegaptanib is not metabolized by the P450 system.26 The drug is eliminated primarily in the urine. Rabbit models showed evidence of 2-fluorouridine, a metabolite of pegaptanib, in the urine and plasma after administration.25 Research suggests that a creatinine clearance of <20 mL/min is associated with a 2.3 times increase in the levels of the drug by AUC calculations.26 However, pegaptanib has been studied in patients with mild to severe renal impairment, and no dost adjustments are needed with the FDA-recommended 0.3-mg dose.25
Ranibizumab
Ranibizumab (Lucentis, Genentech, CA, USA) is a recombinant humanized monoclonal immunoglobulin IgG1 K-isotype antibody fragment, which binds to the receptor-binding domain of all VEGF isoform, which fully inhibits the protein-receptor complex from being formed, and, therefore, stops all downstream signaling. Ranibizumab was derived by affinity maturation of a humanized Fab fragment of the original monoclonal anti-VEGF antibody and has only one antigen-binding site (Fig. 13.3).27 Ranibizumab is a 48-kDa antibody. Its pH is 5.5 (drug insert).
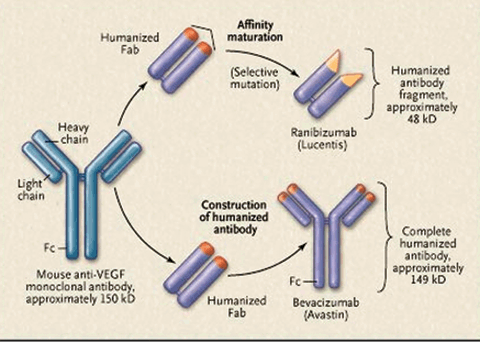
FIGURE 13.3. Relationship between ranibizumab and bevacizumab. Ranibizumab is a recombinant humanized monoclonal IgG1 kappa-isotype antibody fragment (with a molecular weight of about 48 kD). It is produced in an Escherichia coli expression system (and thus is not glycosylated) and is designed for intraocular use. Bevacizumab is a recombinant humanized monoclonal IgG1 antibody (with a molecular weight of about 149 kD). It is produced in a Chinese-hamster-ovary mammalian-cell expression system (and thus is glycosylated) and is designed for intravenous infusion. Both the antibody fragment and the full-length antibody bind to and inhibit all the biologically active forms of VEGF A and are derived from the same mouse monoclonal antibody. However, ranibizumab has been genetically engineered through a process of selective mutation to increase its affinity for binding and inhibiting the growth factor. The Fab domain of ranibizumab differs from that of bevacizumab by six amino acids, five on the heavy chain (four of which are in the binding site) and one on the light chain. Not all the intermediate Fabs between the mouse monoclonal antibody and ranibizumab are shown. (Reproduced from Steinbrook R. N Engl J Med. 2006;355:1409–1412, with permission.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


