Neovascular-AMD Currently in Clinical Trials
Mohamed Ibrahim • Roomasa Channa • Diana V. Do • Quan Dong Nguyen
INTRODUCTION
The prevalence of advanced stages of age-related macular degeneration (AMD) in the United States is estimated to be 1.75 million in 2004 and projected to increase to 3 million by 2020.1 Rein et al. developed a model that predicted substantial increases in the numbers of patients with early and advanced AMD by the year 2050. However, they also showed that currently available treatment options, if used fully, could reduce the vision loss associated with AMD by up to 35%.2 Such encouraging prediction makes a strong case for developing more effective medications to combat vision loss due to AMD.
Among the different treatment modalities for neovascular AMD (NV-AMD), pharmacologic treatments with agents that target vascular endothelial growth factor (VEGF) have shown considerable efficacy in improving visual acuity in patients with AMD.
The VEGF pathway is initiated when VEGF binds to its receptors on endothelial cells. The two best-characterized VEGF receptors are termed VEGF receptor 1 (VEGFR1) and VEGF receptor 2 (VEGFR2). VEGFR1 and VEGFR2 are highly related transmembrane tyrosine kinases that use their ectodomains to bind VEGF; this binding activates the intrinsic tyrosine kinase activity of their cytodomains, initiating intracellular signaling. Interestingly, although VEGFR1 binds to VEGF with substantially higher affinity, most of the biologic effects of VEGF seem to be mediated via VEGFR2.3
In addition to VEGF-A, there are other proteins with similar homology and activities, and they can activate VEGFR1 or VEGFR2. The genes that code for VEGF-A, and the other proteins: VEGF-B, C, and D, and placental growth factors 1 and 2, constitute the VEGF gene family. The role of VEGF family members other than VEGF-A in ocular neovascularization (NV) has not been completely elucidated, but there is evidence to suggest that placental growth factor 1 participates.4
In animals, blockade of the VEGF pathway has been achieved by various means, including blocking antibodies targeted against VEGF5–7 or its receptors8, soluble decoy receptors that prevent VEGF from binding to its normal receptors9–13, as well as small molecule inhibitors of the tyrosine kinase activity of the VEGFRs.14–16 Currently, in clinical trials are several pharmacological agents (Table 11.1) that target various mechanisms of angiogenesis (Fig. 11.1).
Table 11.1.
Antiangiogenic agents currently in clinical trials
|
|
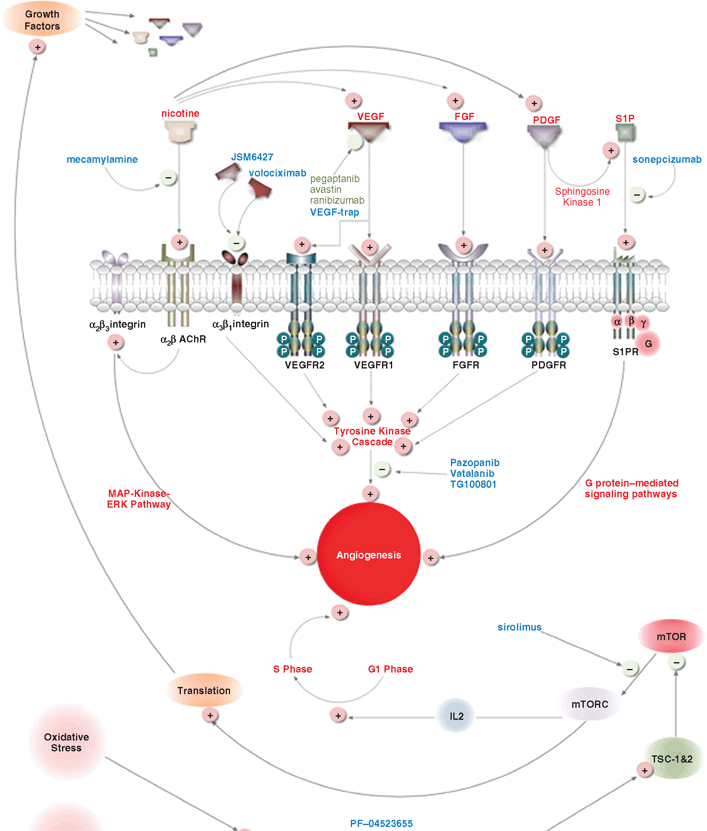
FIGURE 11.1. Pathways of angiogenesis and mechanisms of action of novel antiangiogenic agents currently in clinical trials for NV-AMD.
Pegaptanib, bevacizumab, and ranibizumab are examples of blocking antibodies targeted against VEGFR. Ranibizumab is currently the gold standard for managing choroidal neovascularization (CNV). Some studies have found that one of the most effective ways to block the VEGF signaling pathway is to prevent VEGF from binding to its normal receptors by administering decoy VEGFRs.9,11,17,18 VEGF-Trap is a VEGF-targeting agent that acts as a decoy VEGFR and is currently undergoing phase 3 clinical trials. Vatalanib, pazopanib, and TG108001 are tyrosine kinase inhibitors that target the post-receptor signaling cascade of the VEGFR. Volociximab and JSM6427 are integrin inhibitors that target the α5β1 integrin, which indirectly inhibits the VEGF pathway through inhibition of the post-VEGFR tyrosine kinase cascade.
VEGF pathway is not the only pathway that induces angiogenesis (Fig. 11.1); the process of angiogenesis develops as a result of collaboration of several factors that use different and intertwining pathways. Targeting these angiogenic factors and/or their pathways may provide alternative ways to effectively manage NV-AMD. Alternative methods of blocking angiogenesis include pigment epithelium–derived factor, bioactive lipids, nicotine receptor antagonists, and mTOR inhibitors. Different agents that employ these alternative pathways are currently in different phases of clinical trials in patients with NV-AMD (Table 11.1).
VEGF PATHWAY INHIBITORS
VEGF RECEPTOR DECOY—VEGF TRAP (REGENERON PHARMACEUTICALS)
The parenteral VEGF Trap is a soluble decoy receptor created by fusing the first three Ig domains of VEGFR1 to the constant region (Fc portion) of human IgG1, resulting in a forced homodimer that has picomolar binding affinity.9,11 In tumor experiments, this VEGFR1-Fc reagent is efficacious at approximately 500-fold lower concentration than a similar VEGFR-2 construct.18 Despite this high affinity, the VEGFR1-Fc was not feasible for clinical use because of its poor pharmacokinetic profile. In rodent studies, this protein had to be administered frequently and at very high levels to achieve clinical efficacy.9,11,17 In addition, the VEGFR1-Fc exhibited certain toxicological side effects that are not seen with the VEGFR2-Fc.18
In 2002, VEGF-TrapR1R2 was developed by Holash and colleagues by fusing the second Ig domain of VEGFR1 with the third Ig domain of VEGFR2. VEGF-TrapR1R2 has a very high affinity for VEGF, prolonged in vivo pharmacokinetics and pharmacodynamics, lacks nonspecific toxicities, and can effectively suppress the growth and vascularization of a number of different types of tumors in vivo.3
The receptor portion of the VEGF Trap molecule has a high affinity for all VEGF-A isoforms (Kd < 1 pmol/L), placental growth factors 1 and 2, and VEGF-B, VEGF-C, and VEGF-D.19 Therefore, VEGF Trap is distinguished from ranibizumab by its higher-binding affinity for all VEGF-A isoforms and its ability to inhibit other VEGF family members.19
Intravitreal administration of VEGF Trap strongly suppressed laser-induced CNV in mice20 and primates (Wiegand et al. ARVO abstract 1411, 2005). These findings led to the development of a formulation for intraocular delivery, VEGF Trap-Eye, a formulation using ultrapurified VEGF Trap with a combination and concentration of buffers compatible with ocular tissues. In primate toxicology studies, there were no systemic safety signals after intraocular injections of VEGF Trap-Eye, and there was an excellent ocular safety profile based on ocular examinations, color photography, fluorescein angiography (FA), electroretinography, and postmortem microscopic examination of ocular tissues (unpublished data). The only abnormality identified was mild, reversible inflammation in the anterior chamber and vitreous in some primates after intraocular injection.21
A phase 1 randomized, multicenter, placebo-controlled clinical trial investigating the effect of intravenous VEGF Trap in patients with NVAMD showed elimination of approximately 60% of excess retinal thickness after either single or multiple infusions.19 The maximum tolerated dose of intravenous VEGF Trap in this study population was 1.0 mg/kg. At 3 mg/kg, hypertension and proteinuria, which are side effects associated with systemic anti-VEGF therapy, were noted. The potential risks associated with the intravenous injection of VEGF Trap led to investigation of alternative routes of delivery. VEGF Trap-Eye is an intravitreal formulation, designed to increase the therapeutic window and decrease the adverse events associated with VEGF Trap.
The intravitreal injection of VEGF Trap-Eye was investigated in a phase 1 dose-escalation multicenter clinical trial that was designed to determine the safety, tolerability, maximum tolerated dose, and bioactivity of a single intravitreal injection of VEGF Trap-Eye.21
Twenty-one patients with NV-AMD lesions ≤12 disc areas in size and ≥50% active CNV) with best-corrected visual acuity (BCVA) ≤20/40 received a single intraocular injection of one of the following dosages: 0.05 mg (n = 3), 0.15 mg (n = 3), 0.5 mg (n = 3), 1 mg (n = 6), 2 mg (n = 3), or 4 mg (n = 3) of VEGF Trap-Eye. There were no serious adverse events and no identifiable signs of intraocular inflammation. The mean decrease in excess foveal thickness for all patients was 104.5 μm at 6 weeks, and the mean increase in visual acuity was 4.43 letters. In the two highest dose groups combined (2 and 4 mg), the mean increase in BCVA was 13.5 letters, with three of six patients demonstrating improvement of ≥3 lines and three patients requiring no adjunctive treatment of any type to 12 weeks. Some showed elimination of fluorescein leakage and reduction in area of CNV (Fig. 11.2).21
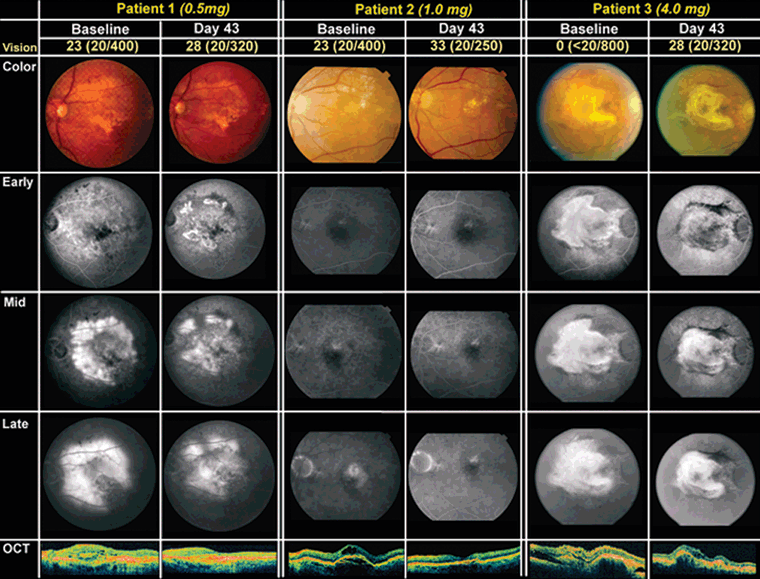
FIGURE 11.2. Changes on fundus photography, FA, and optical coherence tomography of three patients who were participating in the CLEAR-IT 1 Study of intravitreal VEGF Trap-Eye for NV-AMD. Each patient received a single intravitreal injection of VEGF Trap-Eye at baseline at one of several doses (as indicated). Changes were captured and noted at Day 43. (Reprinted with permission from Nguyen, et al. Ophthalmology. 2009;116:2141–2148 e1.)
In a phase 2 double-masked, prospective, randomized, multicenter clinical trial (CLEAR IT-2 Study), 157 patients were randomized to five dose groups and treated with VEGF Trap-Eye in one eye. Two groups initially received monthly doses of 0.5 or 2.0 mg of VEGF Trap-Eye (at weeks 0, 4, 8, and 12), and three groups received quarterly doses of 0.5, 2.0, or 4.0 mg of VEGF Trap-Eye (at baseline and week 12). Following the initial 12-week fixed-dosing phase of the trial, patients continued to receive therapy at the same dose on a PRN dosing schedule based upon the physician’s assessment of the need for re-treatment in accordance with prespecified criteria. Patients were monitored for safety, retinal thickness, and visual acuity. The primary endpoint results of the CLEARIT-2 Study have been reported at various national and international meetings. In this study, VEGF Trap-Eye was generally well tolerated and there were no drug-related serious adverse events. There was one reported case of culture-negative endophthalmitis/uveitis in the study eye, which was deemed not to be drug related. The most common adverse events were those typically associated with intravitreal injections. Patients receiving monthly doses of VEGF Trap-Eye of either 2.0 or 0.5 mg for 12 weeks followed by PRN dosing achieved mean improvements in visual acuity versus baseline of 9 (p < 0.0001) and 5.4 letters (p < 0.085), respectively, at the end of 1 year. The proportion of patients with visual acuity of 20/40 or better increased from 23% at baseline to 45% at week 52 in patients initially treated with 2.0 mg monthly and from 16% at baseline to 47% at week 52 in patients initially treated with 0.5 mg monthly. During week 12 to week 52 PRN dosing period, patients initially dosed on a 2.0 mg monthly schedule received, on average, only 1.6 additional injections and those initially dosed on a 0.5 mg monthly schedule received, on average, 2.5 injections. Patients receiving monthly doses of VEGF Trap-Eye of either 2.0 or 0.5 mg for 12 weeks followed by PRN dosing also achieved mean decreases in retinal thickness versus baseline of 143 μm (p < 0.0001) and 125 μm (p < 0.0001) at week 52, respectively.
VEGF Trap-Eye was also associated with a reduction in the size of the total active CNV. Patients initially receiving either a 2 or 0.5 mg monthly fixed dose of VEGF Trap-Eye for 12 weeks followed by PRN dosing experienced statistically significant 3.41 and 1.42 mm2 reductions in mean CNV size at 48 weeks (the final 1-year analysis from the independent reading center) versus baseline, respectively. Patients in the 2 mg monthly cohort also achieved a statistically significant 1.75 mm2 reduction in total lesion size. A reduction in total lesion size was not seen in the cohort initially dosed with 0.5 mg monthly.
Patients receiving fixed dosing of VEGF Trap-Eye at baseline and week 12 of either 4.0, 2.0, or 0.5 mg followed by PRN dosing achieved mean improvements in visual acuity versus baseline of 4.2 letters (p < 0.015 vs. baseline), 5.2 letters (p < 0.041 vs. baseline), and 2.6 letters (p < 0.344 vs. baseline) respectively, at the end of 1 year. The proportion of patients with visual acuity of 20/40 or better increased from 16% at baseline to 36% at week 52, from 10% at baseline to 45% at week 52, and from 22% at baseline to 34% at week 52, in patients initially treated with 4, 2, and 0.5 mg, respectively. While PRN dosing following a fixed dosing at baseline and week 12 also yielded improvements in visual acuity and retinal thickness versus baseline at week 52, the results generally were not as robust as those obtained with initial fixed monthly dosing.
In an extension to the CLEAR IT-2 study, 117 patients from all groups have been enrolled into one group receiving PRN 2 mg of VEGF Trap-Eye every 2 months. In this extension study, the reported systemic serious adverse events included three deaths, a case of hemorrhagic stroke, and a case of myocardial infarction; all deemed not to be related to the study drug. Compared to baseline, there was an average gain of 7.1 letters at month 18 (p < 0.0001), compared to an average gain of 8.4 letters at month 12. Only 7% showed loss of ≥ 15 letters, 74% either gained vision or maintained their vision at baseline, and 26% gained ≥ 15 letters. The patients who have a vision of ≥20/40 at month 18 were 45% compared to 15% at baseline. During the PRN phase (month 3–18), the mean number of reinjections was 3.5 ± 2.5 (range 0–12), the mean number of days that elapsed before the first PRN injection was administered was 155 ± 130 (range 21–448), and the mean number of days between the first and the second PRN injection was 99 ± 67 (range 22–350). Overall, 50% of the patients needed ≤3 additional injections after month 3 and 75% needed ≤4 injections after month 3.
Regeneron and Bayer HealthCare have joined forces and initiated a global development program for VEGF Trap-Eye in NV-AMD. In two parallel phase 3 trials, which have completed recruitments, the companies are evaluating VEGF Trap-Eye administered at the following dosages: 0.5 mg every 4 weeks, 2 mg every 4 weeks, or 2 mg every 8 weeks (following three monthly doses) in direct comparison with ranibizumab (Lucentis; Genentech, Inc.) administered 0.5 mg every 4 weeks according to its U.S. label during the first year of the studies. In the studies, PRN dosing will be evaluated during the second year of each study. The VIEW1 study enrolled patients in the United States and Canada and the VIEW2 study encompassed patients in Europe, Asia Pacific, Japan, and Latin America (Box 11.1).
BOX 11.1 VEGF Trap-Eye Trials in Wet Macular Degeneration
- CLEAR-IT-2: A randomized, double-masked, active controlled phase 2 Study of the efficacy, safety, and tolerability of repeated doses of intravitreal VEGF trap in subjects with NV-AMD
- Purpose: A phase 2 double-masked, prospective, randomized, multicenter clinical trial to study five different doses of VEGF Trap Eye in patients with neovascular ARMD.
- Method: Two groups initially received monthly doses of 0.5 or 2.0 mg of VEGF Trap-Eye (at weeks 0, 4, 8, and 12) and three groups received quarterly doses of 0.5, 2.0, or 4.0 mg of VEGF Trap-Eye (at baseline and week 12). Following the initial fixed dose schedule, patients received PRN dosing as determined by the physicians.
- Conclusions: Patients receiving monthly doses of VEGF Trap-Eye of either 2.0 or 0.5 mg for 12 weeks followed by PRN dosing achieved mean improvements in visual acuity versus baseline of 9 and 5.4 letters, respectively, at the end of 1 year, and achieved mean decreases in retinal thickness versus baseline of 143 and 125 μm at week 52, respectively.
- CLEAR-IT Extension Study: The extension study showed that patients achieved and maintained significant improvement in visual acuity for 18 months with initial fixed dosing followed by 15 months of PRN. The extension study demonstrates that patients with NV-AMD achieved and maintained significant improvement in BCVA for 18 months with initial fixed dosing followed by 15 months of as-needed administration. On average, only 3.5 injections were given during the PRN dosing phase. Patients continue to be seen and treated in the extension stage of the CLEAR-IT 2 study dosing. On average, only 3.5 injections were given during the PRN dosing phase.
- CLEAR-IT-3: A phase 3 trial is ongoing.
- VIEW Study: VEGF Trap-Eye: Investigation of Efficacy and Safety in Wet AMD
- Purpose: Two parallel phase 3 trials are evaluating VEGF Trap-Eye administered in neovascular ARMD patients at the following dosages: 0.5 mg every 4 weeks, 2 mg every 4 weeks, or 2 mg every 8 weeks (following three monthly doses) in direct comparison with ranibizumab administered 0.5 mg every 4 weeks during the first year. In the studies, PRN dosing will be evaluated during the second year of each study. The VIEW1 study enrolled patients in the United States and Canada, and the VIEW2 study enrolled patients in Europe, Asia Pacific, Japan, and Latin America.
- Status: Ongoing study.
Tyrosine Kinase Inhibitors
Pazopanib (GlaxoSmithKline, Inc.)
Pazopanib (also known as GW786034) is a tyrosine kinase inhibitor that blocks VEGFR receptor (VEGFR)-1, 2, 3, platelet-derived growth factor receptor (PDGFR), c-kit receptor, fibroblast growth factor receptor 1 (FGFR1), FGFR3, and c-fms (Fig. 11.1). By blocking the post-receptor cascade, pazopanib theoretically provides a widespread inhibition of angiogenesis. It has varying affinities for different receptors:
VEGFR1≈VEGFR2≈VEGFR3>PDGFRβ≈PDGFRα≈ cKit>FGFR1≈FGFR3≈cfms>>other kinases.22
Pazopanib was initially developed as an anticancer drug but has been found to inhibit ocular angiogenesis as well. Takashi and colleagues demonstrated that in mouse models, orally administered pazopanib resulted in 93% decrease in the size of CNV lesion compared with vehicle treatment. Periocular injection in similar models resulted in 40% regression in size of CNV, showing that the drug is effective in both locally and systemically administered forms. Another interesting point to note is that pazopanib not only suppresses further growth but also causes regression of the existing CNV lesion.
Pazopanib is being evaluated in several clinical trials as a potential therapeutic agent in patients with NV-AMD. A phase 1 trial designed to establish the safety and tolerability of pazopanib eye drops in humans has been completed on 38 healthy volunteers and elderly subjects with NV-AMD.23 The trial was a two-part study that assessed the tolerability of single and repeat eye drop dosing. A phase 2 study to evaluate the pharmacodynamics, safety, and pharmacokinetics of pazopanib eye drops in patients with NV-AMD will begin in 2010.24,25
Vatalanib (Novartis, Inc.)
Vatalanib (also known as CGP 79787, CGP-797870, PTK/ZK, PTK787, UNII-5DX9U76296, ZK-232934) is an orally administered tyrosine kinase inhibitor. It causes widespread inhibition of multiple VEGFR tyrosine kinases—VEGFR1 (sFlt-1), VEGFR2 (KDR), and VEGFR3 (Flt-4).26
Vatalanib has been studied in a number of preclinical studies and has shown promising results. Daily subcutaneous injections of vatalanib in murine models of retinal NV showed significant inhibition of growth of the lesion.27 Another model showed reduction in angiogenic changes when a single intravitreal injection was administered.28 Oral administration also worked well to inhibit CNV29 and NV in the subretinal space.27 The drug has been used orally in patients with hematologic and solid malignancies; adverse events including fatigue, diarrhea, hypertension, nausea/vomiting have been reported. However, no serious adverse events have been noted with the use of vatalanib thus far.30
Clinical safety and efficacy of oral Vatalanib used in combination with verteporfin (Visudyne; Novartis, Inc) or ranibizumab are being evaluated in patients with subfoveal CNV secondary to AMD in a randomized, double-masked, multicenter, phase 1/2 clinical trial (ADVANCE). The study enrolled 50 patients and initially started with three cohorts: Visudyne + PTK787, 500 mg/day; Visudyne + PTK787, 1,000 mg/day; Visudyne + placebo. The protocol was then modified to use ranibizumab with PTK787 for participants in the second cohort so as to reflect the current standard of care for AMD patients. The primary ouwtcome measure was to assess safety of vatalanib in patients with AMD. Assessment of change in visual acuity, macular edema, and fluorescein leakage from baseline were secondary outcome measures.31
TG100801 (TargeGen Inc.)
TG100801 is an inactive prodrug, which generates the active molecule TG100572 by deesterification. TG100572 inhibits Src kinases and VEGF and PDGFR kinases. Topical formulation is available and is believed to reach the site of action by penetration through the sclera.32 Systemic delivery of TG100572 in a murine model of laser-induced CNV caused significant suppression of CNV but with an associated weight loss suggestive of systemic toxicity. To minimize systemic exposure, topical delivery of TG100572 to the eye was explored, and while substantial levels of TG100572 were achieved in the retina and choroid, higher levels were achieved using TG100801. TG100801 is sufficiently small to cross the sclera enabling an effective topical formulation to be developed. Topical TG100801 significantly suppressed laser-induced CNV in mice, reduced fluorescein leakage from the vasculature and retinal thickening measured by optical coherence tomography in a rat model of retinal vein occlusion. The data suggested that TG100801 may provide a new topically applied treatment approach for ocular NV and retinal edema.32
A phase 1 trial using TG10081 on 42 healthy volunteers has been completed. Preliminary results suggest that the drug is safe for topical administration in human eyes when used twice a day for 14 days.33
Unfortunately, the phase 2 multicenter clinical trial of TG100801 in patients with NV-AMD was discontinued because of keratopathy.33,34
Integrin Inhibitors
JSM6427 (Jerini Ophthalmic, Inc.)
Integrins are an important family of cell surface receptors composed of α-β heterodimers. They play important roles in intercellular adhesion, adhesion to the extracellular matrix, and transmitting signals between cells and from the extracellular matrix.35,36 Different combinations of α-β heterodimers have specific functions in different tissues.36
Integrins are believed to be involved in angiogenesis via transmitting signals from the extracellular matrix, and as downstream effectors of growth factors like VEGF, FGF, and others.36,37 Angiogenic growth factors like FGF and TGF are associated with upregulation of integrin α5β1.38,39
Campochiaro and colleagues showed that integrin α5β1 is upregulated in mouse models of CNV and plays an important role in its development and maintenance.36 Furthermore, they demonstrated that systemic administration of JSM6427, a highly selective competitive antagonist of integrin α5β1, successfully inhibited development of new and promoted regression of established CNV. However, in this study, they were unable to show significant inhibition of CNV following intravitreal administration of JSM6427. The authors suggested that sustained intravitreal drug levels are probably required to provide sufficient inhibition. Endothelial cells in normal vessels are not dependent on α5β1; hence, JSM6427 can provide specific inhibition of pathological CNV.36 The specificity of the drug for pathological vessels suggests a good safety profile.
A phase 1, open-label study using single and repeated intravitreal dosing of JSM6427 was recruiting patients with subfoveal NV due to AMD. The trial was using the drug in four different concentrations: 1.5, 3, 7.5, or 15 mg/mL. The primary outcome measure was to determine the maximum tolerated dose. Other outcome measures included the pharmacokinetics of JSM6427 following single and multiple intravitreal injections and preliminary data on efficacy.13 Due to various reasons, the sponsor, Jerini Ophthalmic, Inc., had to discontinue the study. Hopefully, JSM6427 will be evaluated in other studies in the future.
Volociximab (Ophthotech, Inc.)
Volociximab is a chimeric monoclonal IgG4 antibody that binds α5β1 integrin. It was developed for the treatment of solid tumors and is being tested for the treatment of CNV by Ophthotech Corporation. The molecule has been demonstrated to inhibit endothelial cell proliferation and angiogenesis in in vitro assays. When administered by intravitreal injection concurrently with laser-induced Bruch membrane rupture and then weekly, it inhibits the development of laser-induced CNV in cynomolgus monkeys.40
A phase 1 dose-escalating trial of intravitreal volociximab in combination with ranibizumab is currently recruiting subjects. The trial has aimed to enroll 50 subjects to establish the safety, tolerability, and pharmacokinetics profile of intravitreal injection of volociximab in subjects with NV-AMD. The primary safety end point aims at determining the presence of any dose-limiting toxicity within a 2-year time frame.
PIGMENT EPITHELIAL–DERIVED FACTOR
Pigment epithelial–derived factor (PEDF) is a protein normally synthesized by the retinal pigment epithelium. It has been identified as an important inhibitor of angiogenesis41—ischemic cultured cells are associated with lower levels of PEDF; PEDF levels also have been reported to be lower than normal in ocular diseases like proliferative diabetic retinopathy and CNV.42,43 PEDF inhibits angiogenesis via various mechanisms, which include inducing apoptosis of endothelial cells44,45 and downregulating VEGF expression.46 Furthermore, PEDF is a neuroprotective agent and part of its clinical efficacy may be due to its protective effect on the neurosensory retina.
An E1-, partial E3-, and E4-deleted adenovirus vector was developed to carry the PEDF and formulate the drug—ADGVPEDF.11 (GenVec, Inc.). Intravitreal and subretinal injections of the recombinant adenoviral vector in three different animal models were able to achieve concentrations of PEDF that were sufficiently high to inhibit CNV.47 Further preclinical studies showed that injections were able to cause regression of established CNV.48 Although recognized as an antiangiogenic agent at physiological levels, PEDF has been reported to have proangiogenic effects when used at high doses in laser-induced CNV models in mice. The authors suggested that caution should be exercised when using this agent in dose-escalation clinical trials and also mentioned that drug delivery via an adenoviral vector may not produce the same effect.49
The safety of AdPEDF in 28 patients with NV-AMD was tested in a phase 1, dose-escalation, multicenter clinical trial as the first study employing gene therapy in NV-AMD. The drug was administered via intravitreal injection. Eight dose levels ranging from 10(6) to 10(9.5) particle units were tested. Safety of the injection was closely monitored in thirteen visits conducted over a year. The injection was well tolerated by most patients, with only minimal transient inflammation noted in a few.50 Three to six months after injection 50% of the patients treated with 10(6) to 10(7.5) PU and 94% and 71% of those treated with 10(8) to 10(9.5) PU had either stabilization or decrease in size of lesion. In the high-dose group, the effect of the drug on the lesion was sustained at 12 months after injection, when the authors reported a median increase in lesion size of zero disc areas.50 No serious drug-related adverse effects51–53 have been reported thus far among the patients in this phase 1 study.
BIOACTIVE LIPIDS
There is increasing recognition that many classes of lipids, especially metabolites of cell membrane components, have a role in signal transduction and in regulation of multiple cellular processes. Lipids with such roles are known as “bioactive lipids.” The sphingolipids, formed from the condensation of serine and palmitate, are of particular interest in the regulation of the vascular endothelium. The sphingolipid, sphingomyelin, is an important component of cellular membranes. It is synthesized from and broken down to ceramide, which in turn is broken down by ceramidase to yield sphingosine, and then phosphorylated by the sphingosine kinases (SK1 and SK2) to yield sphingosine-1-phosphate (S1P). S1P is the ligand for five different cell-surface S1P receptors. S1P receptors and SKs have all been shown to be present in the eye.54
S1P is intimately involved in regulation of endothelial function. S1P1 receptors are required for recruitment of vascular smooth muscle cells and pericytes during mouse embryonic vessel formation.55 S1P induces proliferation and migration of human endothelial cells56 and regulates basal and angiopoietin-1–mediated endothelial permeability.57 There is extensive cross talk between S1P and vascular endothelial cell growth factor receptors: S1P can activate VEGFR-2 in the absence of VEGF; VEGF sensitizes endothelium to the action of S1P by upregulating its receptors; activated PDGF receptor activates SK1 on the cell membrane, activating S1PR1. S1P has a role in animal models of retinal disease. In mice, an anti-S1P monoclonal antibody inhibits the development of laser-induced CNV.54
S1P2 receptor knock-out mice demonstrate reduced angiogenesis in the oxygen-induced retinopathy model.58 Systemic treatment with a small molecule inhibitor of SK significantly reduced retinal fluorescein leakage in diabetic rats.59
Sonepcizumab (iSONEP; Lpath, Inc.), is a humanized monoclonal antibody that specifically binds S1P. Intraocular injection of sonepcizumab significantly reduced macrophage influx into ischemic retina and strongly suppressed retinal NV in mice with oxygen-induced ischemic retinopathy. In mice with laser-induced rupture sites in Bruch membrane, intraocular injection of sonepcizumab significantly reduced the area of CNV and concomitantly reduced fluorescein leakage from the remaining CNV. Four weeks after intraocular injection of up to 1.8 mg of the sonepcizumab in nonhuman primates, electroretinogram and fluorescein angiograms were normal, and light microscopy of ocular sections showed no evidence of structural damage. Data collected from animal studies show that S1P stimulates both choroidal and retinal NV and suggest that sonepcizumab could be considered for evaluation in patients with choroidal or retinal NV.60
A monoclonal antibody against S1P has been humanized and is in phase 1 trials as an antiangiogenic agent in cancer treatment. A phase 1 dose-escalating trial is ongoing to evaluate the safety and the preliminary efficacy of single intravitreal injection of five different doses of iSONEP in 15 subjects with NV-AMD. The tested doses range from 0.2 to 1.8 mg, and the subjects will be followed for 12 months.
NICOTINE RECEPTOR ANTAGONISTS
ATG003 (CoMentis, Inc.) is the ophthalmic formulation of the drug mecamylamine. Mecamylamine is a nonspecific antagonist of the nicotine receptor. Nicotine is an alkaloid present in plants such as tobacco and coca. It is fat soluble and can therefore easily cross cell membranes. It is mainly metabolized in the liver to cotinine and excreted in the kidneys.61 Nicotine binds to acetylcholine receptors (AChRs) located on the cell membrane. Over the last decade, nicotinic receptors have been found to be located in multiple nonneuronal tissues, including human endothelial cells.62 Kiuchi and colleagues demonstrated in 2008 that nicotine receptors are expressed on retinal and choroidal endothelial cells.63
Nicotine receptors are made up of multiple subunits (α1-α10, β1-β4, γ, δ, and ε). Endothelial cells possess an α7 AChR, which, when stimulated, indirectly leads to activation of the integrin αvβ3 receptor and an intracellular mitogen-activated protein kinase (ERK) pathway that further leads to angiogenesis.64 Specific antagonists of this receptor such as α-bungarotoxin and 65 as well as the nonspecific antagonist mecamylamine have been shown to inhibit proliferation of endothelial cells from different tissues.66
In addition to direct effects mediated via the AChR, nicotine has also been shown to induce angiogenic factors like FGF, PDGF, and VEGF.67,68 It was also found that nicotine increases the expression of VEGF-2 receptor.69 Nicotine increased the size of the laser-induced choroidal neovascular lesions in mice, but the CNV was inhibited by subcutaneous infusion of 50 mg/kg/day of mecamylamine. Moreover, mecamylamine inhibited growth of CNV lesion even in the absence of exogenous administration of nicotine, implying that acetylcholine is responsible for stimulating nicotinic receptors in vivo and promoting growth of CNV. It has been demonstrated that topical drops of 0.1% or 1% mecamylamine can significantly decrease CNV in mice.63 The success of the topical drops in mice led to testing in humans.
Topical formulation of ATG3 (mecamylamine HCl) was evaluated in a phase 1 randomized, double-masked, placebo-controlled study that enrolled 80 healthy volunteers in single and multiple dose ascending regimens for up to 14 days of therapy. Subjects received the eye drops twice daily. There were no study medication-related systemic side effects. Mecamylamine levels in plasma were found to be very low following eye drop application. Two Phase 2 studies, one comparing the safety and efficacy of two formulations of ATG003 (1.0% and 0.3%) versus placebo in patients with neovascular age-related macular degeneration (NV-AMD), and another comparing 1.0% ATG003 versus placebo in patients on treatment with anti-VEGF therapy for NV-AMD have been conducted; the former study has been terminated, the latter is ongoing.70,71
mTOR PATHWAY MODULATORS
SIROLIMUS (MACUSIGHT INC.)
Sirolimus (rapamycin) is a macrolide antibiotic that is produced naturally by Streptomyces hygroscopicus.72 The mechanism of action of sirolimus in immunomodulation has been described extensively in medical literature.73–76 Sirolimus binds to FKBP12 forming a complex that binds to the mammalian target of rapamycin (mTOR), inhibiting its function in the mTORC1 complex.77 mTOR is a key component in the cell’s response to insulin, insulin-like growth factors, nutrient levels, hypoxia, and redox status. mTOR forms the complexes mTORC1 and mTORC2.78 mTORC1 promotes translation by phosphorylating and activating the p70-S6 Kinase 1 (S6K1) and phosphorylating and inactivating eukaryotic initiation factor 4E (eIF4E)-binding protein 1(4E-BP1), which is a repressor of translation.78 The inhibition of mTOR blocks IL-2– mediated signal transduction pathways. Such blockage arrests cell cycle progression from G1 to S phase in T cells, endothelial cells, osteosarcoma cells, myogenic cell lines, and smooth muscle cells.74,79 In addition, sirolimus inhibits the production of antibodies.74,80
Sirolimus’ inhibition of mTOR activity has been shown to impact a number of the components of the angiogenic process. Sirolimus inhibits the production, signaling, and activity of many growth factors reported to be relevant to the development of dry eye syndrome, diabetic retinopathy, and CNV. Moreover, sirolimus can modulate the contribution of progenitor stem cells to CNV, reduce permeability, blunt complement-mediated effects and downregulate genes involved in the inflammatory process.81–87
It is hypothesized that as mTOR plays a critical role in regulating basic cellular functions including cell proliferation, survival, mobility, and angiogenesis, inhibitors of mTOR are expected to show a therapeutic effect in treating dry eye, posterior uveitis, and the pathological angiogenesis of CNV.
Sirolimus underwent phase 1 testing in a randomized, open-label, dose-escalation clinical study. The study aimed at the assessment of safety, tolerability, and preliminary bioactivity of intravitreal or subconjunctival injection of sirolimus in patients with newly diagnosed NV-AMD. The maximum tested doses in this study were 880 and 176 μg for the subconjunctival and the intravitreal routes, respectively.88
A total of 30 patients were enrolled in this study. All patients were followed for 12 months after a single injection of sirolimus. The intravitreal and subconjunctival injections of sirolimus were found to be both safe and tolerable in all tested doses when used either as intravitreal or as subconjunctival injections. In addition, sirolimus showed preliminary bioactivity indicated by improvements in visual acuity that were associated with corresponding reductions in retinal thickness as detected by optical coherence tomography. The preliminary findings demonstrated that sirolimus administered via subconjunctival injections (Fig. 11.3) was as effective as sirolimus administered via intravitreal injections (Fig. 11.4).88 Intravitreal injections, while currently the standard route of administration for AMD therapies, are uncomfortable for many patients and are accompanied by the risk of serious infection in a small percentage of patients. In contrast, subconjunctival injections are designed to offer physicians and patients a less invasive and more convenient procedure.88
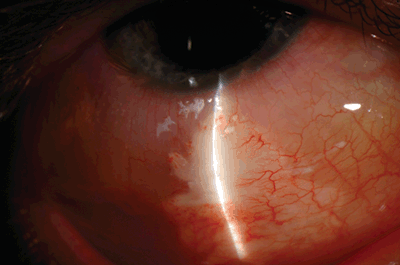
FIGURE 11.3. Slit-lamp image showing white subconjunctival deposits following subconjunctival injection of sirolimus.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


