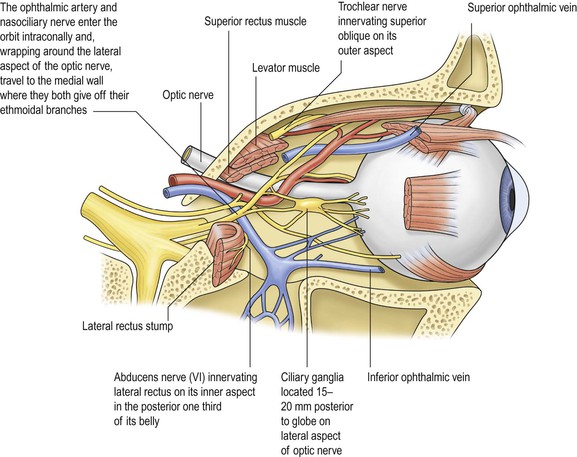
I. The orbital volume is approximately 30 ml, and the orbital depth (anterior to posterior) is approximately 4.5 cm (Figs. 14.1–14.5). A. The medial orbital wall is quite thin (<0.5 mm) and transparent. II. In addition to the bony walls, the eye, and the optic nerve, the orbit contains many soft-tissue structures such as fat, muscle (striated and nonstriated), cartilage, bone, fibrous tissue, nerves, and blood vessels. B. All orbital structures may be involved in disease processes. I. The main clinical manifestation of orbital disease is exophthalmos (ocular proptosis), the extent and direction of which depend on a number of factors1: (1) size of lesion, (2) character of lesion (expansile vs. infiltrative growth, rapid vs. slow growth), (3) location of the lesion in the orbit (small lesion in muscle cone causes more exophthalmos than lesion of same size outside the muscle cone; lesions anterior to septum orbitale do not produce exophthalmos unless they also grow posteriorly), and (4) lesion’s effect on the extraocular muscles (complete paralysis of all muscles by itself can cause 2 mm of exophthalmos). Developmental abnormalities are usually associated with abnormalities of the cranial and facial bones such as tower skull or hypertelorism. I. Nonsuppurative (see section Nonsuppurative, Chronic Nongranulomatous Uveitis and Endophthalmitis in Chapter 3)—orbital cellulitis is most commonly caused by extension of an inflammation from the paranasal sinuses (Fig. 14.6). II. Suppurative (see section Suppurative Endophthalmitis and Panophthalmitis in Chapter 3) A. Purulent infection (e.g., with Staphylococcus) occurs commonly after trauma. B. Phycomycosis (mucormycosis) is a devastating cause of suppurative orbital inflammation (Fig. 14.7; see Chapter 4). B. A rare cause is the benign lymphoepithelial lesion of Godwin2 (Fig. 14.8). 2. It may be part of Sjögren’s syndrome (see later). 3. Rarely, it may become malignant. C. Sjögren’s syndrome (see Fig. 14.8) 2. The tissue destruction results in the symptoms of keratoconjunctivitis sicca and xerostomia. 3. Indirect support exists for a putative role of the Epstein–Barr virus (see Chapter 3) in the pathogenesis of the disease. D. Inflammatory pseudotumor (see later in this chapter) II. Granulomatous A. Granulomatous inflammations rarely involve the orbit. 4. Histologically, a granulomatous reaction surrounds cholesterol crystals and altered blood. I. Direct effect on whatever tissue may be wounded by the injury II. Complications (indirect effect) A. Infection 1. Organisms may be introduced at the time of injury. a. Bacteria cause an acute purulent inflammation. b. Fungi cause a delayed, chronic, granulomatous inflammation. B. Inflammation 1. The inflammation may be secondary to toxic products of tissue destruction. 2. Orbital thrombophlebitis may result. 3. A retained intraorbital foreign body may induce inflammation. The effects of nonpenetrating wounds are those secondary to contusion and concussion, mainly hemorrhage, secondary muscle palsies, and infraorbital nerve involvement. I. Primary orbital vascular disease is rare. II. Causes include varices, arteriovenular aneurysm, thrombophlebitis, and cavernous sinus thrombosis. Orbital varix, also called distensible venous malformation (Fig. 14.9), may occur anterior to the septum orbitale and not cause exophthalmos, or it may occur posterior to the septum orbitale, causing exophthalmos. The exophthalmos may be acute if the varix undergoes thrombosis. I. Classification of eye changes of Graves’ disease (Box 14.1) II. Mild form (“thyrotoxic” exophthalmos) B. It may present initially with unilateral involvement but usually becomes bilateral. C. Clinically and chemically, the patient is hyperthyroid. D. Lid retraction, the most common clinical sign, may simulate exophthalmos. E. Occasionally exophthalmos is present. F. Prognosis for vision is good. III. Severe form (“thyrotropic” or “malignant” exophthalmos; thyroid ophthalmopathy; thyroid orbitopathy) A. The severe form is an autoimmune disease that affects people in middle age (average age, 50 years). 1. The disease is characterized by an increased percentage of suppressor/cytotoxic T lymphocytes. 2. Circulating T cells are directed against thyroid follicular cell antigens. C. Clinically and chemically, the patient may be hyperthyroid, hypothyroid, or euthyroid. E. Prognosis for vision is poor. F. Histologically, the orbital tissue is characterized predominantly by extraocular and periorbital muscle involvement by edema, lymphocytic infiltration (mainly CD4+ and CD8+ T cells along with some focal aggregates of B cells, plasma cells, and mast cells), endomysial fibrosis, and mucopolysaccharide deposition. A. Its onset is between 20 and 30 years of age, with patients rarely living beyond 40 or 50 years. B. Frontal baldness and endocrinopathy, especially testicular atrophy, are common. II. Ocular findings B. Foveal dystrophy that hardly affects vision and pigmentary retinopathy III. Histologically, selective atrophy of muscle fibers is seen. A. Leber’s hereditary optic atrophy (see Chapter 13) B. CPEO 2. Ptosis, external ophthalmoplegia, and often a pigmentary retinopathy can be seen. C. KSS 2. Patients have short stature and increased serum and cerebrospinal fluid lactate levels. 5. Histologically, along with the characteristic ragged-red appearance seen under light microscopy with the modified trichrome stain, electron microscopy shows an increased number of mitochondria containing abnormal cristae. a. The eyes show abnormalities of the macular photoreceptor–retinal pigment epithelium (RPE)–choriocapillaris complex, namely absent or degenerated outer segments and hyperpigmented and hypopigmented RPE. 1) The cytoplasm of RPE cells show ballooned, structurally abnormal (“giant”) mitochondria. See Box 14.2 for classification of neoplasms and other tumors. Box 14.2 Classification of Neoplasms and Other Tumors
Orbit
Normal Anatomy
Exophthalmos
Developmental Abnormalities
Developmental Abnormalities of Bony Orbit
Orbital Inflammation
Acute
Chronic
Injuries
Penetrating Wounds
Nonpenetrating Wounds
Vascular Disease
Primary
Part of Systemic Disease
Ocular Muscle Involvement in Systemic Disease
Graves’ Disease (Fig. 14.10)
Myotonic Dystrophy (Myotonia Dystrophica; Steinert’s Disease)
Mitochondrial Myopathies
Neoplasms and Other Tumors3
Orbit
14