Viral Disease of the Ocular Anterior Segment: Basic Science and Clinical Disease
Deborah Pavan-Langston
VIRUSES
General Characteristics
Unlike bacteria, fungi, and protozoa, viruses are obligate intracellular parasites dependent on the biosynthetic pathways of the host cell for replication. They may infect bacteria, certain other viruses, plants, and animals. This chapter focuses on animal viruses that infect the eye directly or through systemic viral illness.
Classification and Structure
The parasitic nature of viruses is a result of their simple construction: a nucleic acid core of single- or double-stranded RNA or DNA within a protein coat (the capsid). Some viruses, such as the herpesviruses, have their nucleocapsid (core plus capsid) surrounded by a lipoprotein coat, the envelope. This envelope contains components of both host cell membrane and virus-specific protein subunits (Fig. 14-1). This combination confers immunologic immunity and is critical to early infection events. The most commonly used criteria of classification include both physical and biochemical characteristics, such as type of nucleic acid and capsid, size and morphology (e.g., presence or absence of an envelope), as well as tissue and host tropism and means of replication (1, 2, 3, 4).
Virion size is measured in nanometers (nm), with the clinically important viruses ranging from 18 nm (parvoviruses, associated with adenovirus) to 300 nm (poxvirus). The repetitive structural protein pattern of the capsid reduces the amount of viral genome that must be used for encoding the capsid components. Animal viruses have three types of symmetry: icosahedral, helical, and complex symmetry, with the first two being the simplest in structure. The helical viruses have repeating subunits of capsids bound along the helical spiral of the viral nucleic acid core. The icosahedral symmetry is nearly spherical, with multiple axes of rotational symmetry around the core.
Replication
RNA viruses usually synthesize all viral products simultaneously, whereas DNA virus replication is divided into four multistep phases or periods (2, 3, 4, 5). The early phase involves viral recognition of an appropriate target cell, attachment to and penetration of the cell plasma membrane, intracytoplasmic uncoating of the viral nucleic acid, and, with some viruses, delivery of the genome to the nucleus. Once the genome is uncoated in the early phase, infectivity and identifiable structure are lost, thus beginning the eclipse period. This period ends with appearance of new virions after virus assembly. The late phase includes synthesis of early messenger RNA (mRNA) and nonstructural proteins, replication of the genome, late mRNA and structural protein synthesis, posttranslational modification of protein such as phosphorylation, and assembly of new virions (ending the eclipse phase). The latent period includes the eclipse phase and ends with release of virus. One cell may produce 100,000 particles (virions), but only 1% to 10% may be infectious. The noninfectious defective virions result from mutations and errors in manufacture.
Viral recognition of and binding to a host cell is a function of the types of receptors on the cell species (host range), the cell type (tissue tropism), or both. For example, the herpesvirus Epstein-Barr virus (EBV), which causes mononucleosis, has a very limited host range and tissue tropism because it binds to the C3d receptor on human B lymphocytes. The attachment structure for a capsid virus (no envelope) may be part of the capsid or a protein extending from it. The receptors on the cell may be proteins or carbohydrates on glycoproteins or glycolipids. Enveloped viruses have specific glycoproteins as attachment structures.
Most viruses enter the cell by receptor-mediated endocytosis through cell endosomes or by direct injection into the cell through viropexis. After uncoating, the genome of DNA viruses (with the exception of poxviruses) is delivered to the cell nucleus, whereas most RNA viruses remain in the cytoplasm for the entire replicative cycle. Regardless of
whether the infecting nucleic acid is DNA or RNA, the critical step of translation depends on the production of mRNA using the viral genome as the template. The host cell enzymes for making mRNA from a DNA template are all intranuclear and therefore inaccessible to RNA viruses, which are all cytoplasmic. Host cells lack the enzymes to make mRNA from an RNA template. As a result, positivestranded RNA viruses serve as their own mRNA templates. With negative-stranded RNA viruses, such as human immunodeficiency virus (HIV), the RNA polymerase, reverse transcriptase, facilitates the making of positive-stranded mRNA, leading to viral RNA production (2).
whether the infecting nucleic acid is DNA or RNA, the critical step of translation depends on the production of mRNA using the viral genome as the template. The host cell enzymes for making mRNA from a DNA template are all intranuclear and therefore inaccessible to RNA viruses, which are all cytoplasmic. Host cells lack the enzymes to make mRNA from an RNA template. As a result, positivestranded RNA viruses serve as their own mRNA templates. With negative-stranded RNA viruses, such as human immunodeficiency virus (HIV), the RNA polymerase, reverse transcriptase, facilitates the making of positive-stranded mRNA, leading to viral RNA production (2).
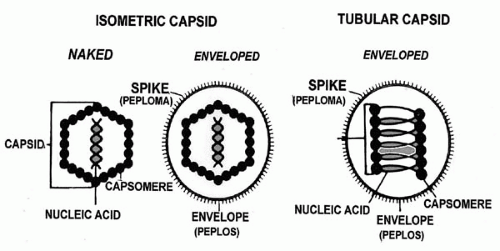 FIGURE 14-1. Diagrams of viral taxonomic groups discussed in this chapter, showing relative size and structure. (Adapted in part from Kinchington P. Virology. In: Smolin G, Thoft R, eds. The cornea, 3rd ed. Boston: Little, Brown 1994:169-183.) |
Virion assembly depends on where the genome replication occurred and whether the final product is a naked capsid or an enveloped virus. Capsid viruses may be assembled around the new genomes or built as empty structures to be filled with the genome later in the cycle. With enveloped viruses, after the nucleocapsid is formed, new viral glycoproteins are sent to cellular membranes by vesicular transport. The envelope is acquired after the nucleocapsid associates with the viral glycoprotein-containing regions of host cell membranes in the budding process. This process may occur at the plasma membrane with release (RNA viruses), in the endoplasmic reticulum en route to the surface (flaviviruses), or at the nuclear membrane with release into the endoplasmic reticulum and transport to the surface. In all cases the virion is released by exocytosis, cell lysis, or by cell-to-cell bridges (herpesviruses) (1, 2, 3, 4).
IMMUNE RESPONSE TO VIRAL INFECTION
Host immune response to viral invasion involves both nonspecific (natural) and specific (acquired) defenses (4,6, 7, 8). The nonspecific category includes the complement system, primitive cellular immunity (macrophages and natural killer cells), and interferon, none of which requires previous exposure to the viral antigens. The interferons, in particular, inhibit virtually all phases of viral infection from penetration to assembly, but interferons may also cause a damaging inflammatory response.
Specific immune responses take a few days to begin and are stimulated by viral proteins that are part of the virus particle or on the surface of infected cells, resulting in B- and T-lymphocyte activation. The viral antigens react with macrophages with type II histocompatibility antigens on the surface, thus precipitating both humoral (B cells) and cellular (T cells) immunity. Neutralizing antibodies bind virus, making it noninfectious. Other antibodies do not affect infectivity but bind the virus and stimulate phagocytosis or the complement system, which result in viral death. Complement may lyse free virus and virus-infected cells in the presence of antibody (4,6, 7, 8).
Specific cell-mediated immunity (CMI) includes activation of cytotoxic T-lymphocytes and delayed-type hypersensitivity T cells by viral proteins on the host cell membrane, not by soluble antigen (6, 7, 8). The cytotoxic T lymphocytes destroy cells and present viral antigen on their cell membrane surfaces. Delayed-type T cells secrete lymphokines chemotactic for macrophages and neutrophils. Helper T cells facilitate both B- and T-cell activity, and suppressor T cells downregulate the antiviral immune response. As is noted later in the discussion of herpetic disease, the immune response may, on occasion, be more destructive than the infectious disease itself.
LABORATORY DIAGNOSTIC TECHNIQUES
The general laboratory methods used to confirm clinical diagnosis include (a) direct-view morphology, (b) immunomorphology, (c) immunologic virology, (d) viral culture, (e) serology, and (f) molecular virology (1, 2, 3, 9,10).
Direct-view morphology includes cytologic testing and electron microscopy. Scrapings are taken directly from the lesions for cytology and smeared on a glass slide, fixed in Bouin’s solution for 1 hour, and stained with Giemsa, Tzanck, hematoxylin and eosin, or Papanicolaou stain before light microscopy examination. This technique is useful for rapid diagnosis of the herpesviruses, herpes simplex virus (HSV), varicella zoster virus (VZV), and cytomegalovirus (CMV), as well as measles and rabies infection; it is not as specific or sensitive as viral isolation or immunotesting. Typical changes include change in cell morphology, cell lysis, vacuoles, syncytia, and inclusion bodies. Syncytia are multinucleated giant cells formed by viral fusion of adjacent cells. The herpesviruses, HIV, and paramyxoviruses induce this fusion. Inclusion bodies are either histologic changes in the cells due to viral components or virus-induced changes in cell structure. Cowdry type A inclusions, seen in HSV and VZV infections, are brilliant, large intranuclear inclusions surrounded by a halo separating them from the nuclear membrane. CMV may induce nuclear owl’s eye inclusions, and adenovirus produces smudgy eosinophilic nuclear inclusions. Molluscum contagiosum virus and poxvirus may cause eosinophilic cytoplasmic inclusions, and rabies may cause Negri bodies, rabies virus inclusions in brain tissue (1, 2, 3,10).
Electron microscopy is not a standard laboratory test but can be very useful if sufficient viral particles (106 to 107) are present, and particularly if virus-specific antibody has been added to the sample to cause viral clumping, thus facilitating detection and identification of the virus (immunoelectron microscopy). Definitive diagnosis often cannot be made, however, because many viruses such as the herpesviruses are indistinguishable by electron microscopy.
Immunomorphology includes the immunofluorescence and immunoperoxidase techniques. Direct, indirect, and anti-complement immunofluorescent staining are used for detection of viral antigens in infected cells. Scrapings are taken from the lesion and smeared on a glass slide. In the direct method, the slide is treated with fluorescein-conjugated virus-specific antibody, and incubated for 30 to 60 minutes before buffered saline washing. Reading is under fluorescent microscopy. This is an easier and faster technique than the indirect, but it is less sensitive. In the indirect method, step one is covering the slide with unlabeled virus-specific antibody, step two washing and applying normal serum to decrease nonspecific antibody binding, and step 3 coating the slide with a fluorescein-labeled, anti-immunoglobulin conjugate. This is more sensitive because more label can be bound to the antigen. Immunofluorescence is routinely used to detect herpesvirus, adenovirus, CMV, mumps, measles, respiratory syncytial virus, and rabies (1,2,10,11).
Immunoperoxidase is a commonly used staining technique that uses antibody conjugated with enzymes such as horseradish peroxidase rather than fluorescein as an indicator. The technique is similar to direct immunofluorescence after normal serum overlay to decrease nonspecific staining. The peroxidase enzyme produces an orangebrown precipitate localized at the binding sites in the specimen. This technique has the advantage of being more permanent and being read under light rather than fluorescent microscopy, thus allowing viewing of adjacent tissues for histopathologic analysis (1,3,10, 11, 12).
Immunologic virology involves enzyme immunoassay (EIA) for the detection of viral antigen on the cell surface or within the cell. The enzyme-linked immunosorbent assay (ELISA), radioimmunoassay, and latex agglutination are used to detect virus or antigen released from infected cells (1,2,10). ELISA tests come in a variety of commercially available formats for detection of several viruses by solid-phase or membrane-phase technique. In the solid-phase test, virus-specific antibody is bound to a solid surface such as a plastic microtiter well or tube. In membrane tests, the solid surface is replaced by a membrane. The specimen is put in and captured by the antibody. It is then detected by binding of an enzyme-linked antibody to the antigen, washing, and adding a colorimetric substrate of the enzyme. Solid-phase tests are best suited for batch testing, whereas membrane tests may be used in single tests with little equipment in the physician’s office to detect HSV types 1 and 2 (differentiating from VZV), CMV, adenovirus, and rubella (13,14). Advances in ELISA methodology include fluorescent and enzymatic labels, antibodycapture formats, and monoclonal antibodies, thus making ELISA a frontrunner in routine serologic assay techniques.
Viral culture and isolation is still the gold standard of diagnostic virology. Tissue culture cells are used to grow viruses. Primary cell cultures are cells trypsinized free from animal tissue and grown in monolayers in tubes or flasks. Diploid cell lines are a single cell type that may be passed in sequential generations several but a finite number of times before dying out. Tumor cell lines originate from patient cancers and are immortalized, being passed indefinitely without senescing. Human fetal diploid cells are fibroblastic and support the culture of a wide variety of viruses, including HSV, VZV, CMV, adenovirus, picornavirus. Hep-2, a continuous cell line from a human cancer, also supports many viruses such as adenovirus, HSV, and respiratory syncytial virus. Primary monkey kidney cells are excellent for growth of myxoviruses, some adenoviruses, and enterovirus (3,10).
After tissue culture inoculation of the clinical specimen, the virus may first be detected with light microscopy by changes in the cell monolayer, called cytopathic effect. This is usually a rounding up and ballooning of infected cells that may pull away from the monolayer to leave open “plaques.” HSV can produce cytopathic effect within 1 to 2 days, whereas CMV, adenovirus, and rubella may take up to 1 month. The virus is identified by virus-specific antibody tests such as immunofluorescence, immunoperoxidase, EIA, or ELISA tests. These techniques may even be used in the pre-cytopathic effect stage to speed up diagnosis (1,13).
Serologic test methods involve both quantitative and qualitative evaluation of humoral immunity and antibody production. This may be done by using specific viral antibodies to identify unknown viral isolates or antigens, as noted in the discussion of viral tissue culture, or antibody may be identified using panels of known antigens or for the quantitation of antibody in the sera of infected patients. Methods used for antibody quantitation, discussed previously or elsewhere in this chapter, include viral neutralization, complement fixation, hemagglutination inhibition, ELISA, Western immunoblotting, immunofluorescence, latex particle agglutination, and gel immunodiffusion. Complement-fixing antibodies are quite transient, whereas neutralizing antibodies are often present for years after infection.
ELISA tests are used for viral antigen detection, but are particularly useful for detection of immunoglobulin M (IgM) in the presence of IgG. Only IgM antibodies, if present in the serum, are bound to the solid phase and therefore easily detectable. This is important diagnostically because IgM antibodies appear early during infection and last only a few weeks, whereas IgG appears after 1 or 2 weeks but lasts for years. Quantitative documentation of a fourfold rise in either IgM or IgG strongly supports the diagnosis. Serum should be drawn as soon as possible in the acute illness and again 2 to 3 weeks later for comparative titers. Finding a positive IgM in a single specimen may also be diagnostic in a very ill patient (e.g. Ebola virus) or a patient with ongoing infection (e.g., HIV). Elevation of IgM may also indicate reactivation of a latent infection. IgM detection is most useful in diagnosis of VZV, EBV, CMV, measles, rubella, coxsackieviruses, and hepatitis. Finding IgM in a newborn is diagnostic of intrauterine infection because IgM does not cross the placental barrier (2,4,10,15).
Viral neutralization testing is of value only if a change in titer can be demonstrated in two samples, acutely and at 3 to 4 weeks, because these antibodies last for years after an infection. Change in titer is shown by adding sequential dilutions of serum to tissue culture growing the virus. The titer is the reciprocal of the highest dilution at which the viral cytopathic effect in inhibited.
Complement fixation testing is based on the fact that complement, a complex of multiple plasma proteins that act sequentially to lyse infected cells, combines with viral antigen only in the presence of virus-specific antibody. Antigen and complement are mixed with the patient’s serum. If complement is bound by the effective complex, there will be no hemolysis when this is mixed with hemolysinsensitized sheep red cells. If complement is free because the antigen and antiserum did not match, there will be hemolysis. The test can be arduous and yield nonspecific results.
Agglutination tests detect viral antigens based on the visible clumping of particles, such as latex, red blood cells, or polystyrene, to which virus-specific antibody has been absorbed. This is not commonly used in ocular disease diagnosis. Agglutination of red blood cells occurs in the presence of vaccinia, adenovirus, rubella, mumps, measles, and Newcastle disease. Acute and convalescent host serum is added to a known viral antigen concentration. If the serum inactivates the virus antigen, there will be no agglutination and diagnosis is made. A diagnosis of primary infection, however, requires a fourfold rise in serum antibody titer between acute and convalescent sera. This test is usually done at reference laboratories.
Molecular virology uses recombinant DNA or RNA technology in highly specific tests to detect viral nucleic acids rather than viral protein antigen. Genetic structure and sequence are the distinguishing factors of the family, type, and strain of virus. The unique electrophoretic patterns of RNA viruses or the DNA fragment lengths obtained through restriction endonuclease treatment of nucleic acid samples yield highly specific and sensitive diagnostic data. DNA probes can be made with sequences complementary to specific regions of a known viral genome. These probes are labeled radioactively or with fluorescein or peroxidase, bind the complementary sites in the viral DNA under investigation, and are then read against known probes and viral nucleic acid by autoradiography or under fluorescent or light microscopy. Genetic sequences can also be detected in fixed tissue by in situ hybridization, thus revealing cellular location of the nucleic acid. Other detection techniques include Southern blot (DNA virus) and Northern blot (RNA virus), in which patient viral genome fragments are blotted onto nitrocellulose filters and then detected on the filter by their hybridization to DNA or RNA/DNA probes with autoradiography or EIA-like methods (1, 2, 3).
Amplification techniques have been developed to further the application and usefulness of this technique. The best known is the polymerase chain reaction (PCR). This technique allows a single copy of a genome to be amplified in vitro more than a millionfold in just a few hours (16). This amplification allows the detection and identification of very small amounts of viral nucleic acids using labeled probes in a hybridization assay. PCR can also be modified to detect viral RNA by use of reverse transcriptase to convert RNA to DNA before running the sample. A new version of this test, the real-time quantitative PCR, is an amplification reaction performed using fluorescent probes or DNA intercalating dyes that increase in fluorescence as it quantifies the pathogen load (17). It has a sensitivity of fewer than 10 organisms for all pathogens. Nucleic acid hybridization is quickly becoming the procedure of choice for diagnostic testing in many laboratories.
CHARACTERISTICS OF THE MAJOR OCULAR VIRAL PATHOGEN FAMILIES
Herpesviruses
Family: Herpesviridae
Subfamilies:
Alphaherpesvirinae
Genus: Simplexvirus (HSV types 1 and 2)
Genus: Varicellovirus (VZV)
Betaherpesvirinae
Genus: Cytomegalovirus
Genus: Roseolovirus
Genus: Human herpesvirus 7
Gammaherpesvirinae (lymphoproliferative)
Genus: Lymphocryptovirus (EBV)
Genus: Human herpesvirus 8 (Kaposi’s sarcoma)
The members of this family include HSV types 1 and 2, VZV, CMV, EBV, human herpesviruses 6 (HHV6, roseola), 7 (HHV7, lymphoproliferative) and 8 (HHV8, Kaposi’s sarcoma). These DNA viruses have several characteristics in common, including morphology, mode of replication, ability to cause lytic disease or establish recurrent and latent infections, ability to encode proteins and enzymes in interaction with the host cell, and CMI as the key to control infection but, in some cases, worsen disease (2, 3, 4).
These large (150 to 200 nm), enveloped viruses contain a linear, double-stranded DNA within an icosadeltahedral capsid that is surrounded by a glycoprotein-containing envelope derived from the host cell membrane (Fig. 14-2). The HSV genome encodes approximately 80 proteins, half of which are needed for viral replication and the others for the virus’ interaction with host cells and an immune response. The glycoproteins are encoded by the virus for purposes of attachment, fusion, replication, and avoiding immune control. The last works by IgG binding to viral gE/gI glycoproteins, thereby camouflaging virus and virus-infected cells and reducing antibody effectiveness.
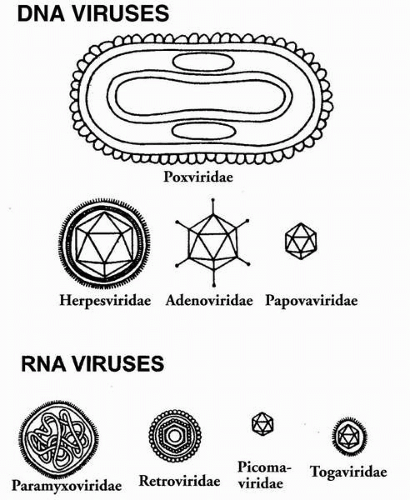 FIGURE 14-2. Diagram showing structural components of naked and enveloped viruses. (Adapted in part from Kinchington P. Virology. In: Smolin G, Thoft R, eds. The cornea, 3rd ed. Boston: Little, Brown 1994:169-183.) |
HSV-1 and HSV-2 are spread by vesicle fluid, saliva, and vaginal secretions. They may infect most types of human and nonhuman cell types, causing lytic infection of epithelial cells and fibroblasts and latent infection in neurons. Cells that support latent infection transcribe only certain genes without genome replication. With reactivation there is progression to early and late gene expression, leading to lytic infection and cell death (3). Triggers of reactivation are not completely understood. In animal models of latency, corneal intrastromal injection, neurosurgical disturbance of the fifth cranial nerve, iontophoresis, and corneal trauma may all induce reactivation, but the final common pathway for this reactivation is unknown. The resulting clinical disease may vary in severity depending on neurovirulence and pathogenicity, and variations in host system response (4,18,19).
Transcription of the viral genome and protein synthesis proceeds in three phases: (a) immediate early protein (alpha) gene transcription through DNA-binding proteins; (b) early proteins (beta) producing more transcription factors and enzymes such as DNA polymerase and thymidine kinase (TK); and (c) late proteins (gamma) with production of structural proteins triggered by genome replication. The remainder of reproduction proceeds as described previously in the section on Replication. HSV production of TK in the beta phase is a critical factor that makes it susceptible to several antiviral agents. If the infection is to be a latent one (i.e., neuronal), the only region of the genome transcribed generates the latency-associated transcripts. These RNAs are not translated into protein and replication does not proceed until there is viral reactivation (3,19).
VZV primary infection manifests as chickenpox, which is acquired through the respiratory tract and disseminated by viremia, ultimately becoming latent in the sensory ganglia. As reactivated disease, it is zoster or shingles. The virus is morphologically identical to HSV but, unlike the latter, there is no reported antigenic variation among VZV strains (20,21). VZV and HSV share several minor antigens, at least six glycoproteins, replicative pathways, the ability to establish latent neuronal infection and recurrent disease, characteristic blister-like skin and mucous membrane lesions, the encoding of TK that makes it susceptible to several antiviral drugs, and control of infection by CMI (2,3,5). The glycoproteins are potent inducers of antibody and may be an integral part of HSV and VZV vaccines in the future (22). These herpesviruses also share a role as the most common infectious causes of anterior segment visual
loss in the developed world (23). VZV is fastidious and hard to recover from clinical samples. Human fibroblast lines are best for tissue culture recovery, and likelihood of recovery is increased by culturing vesicular fluid located around the eye or on the V-1 dermatome. Cytopathic effect appears between 5 and 28 days in culture. Because of this difficulty, VZV is usually identified by immunofluorescence, viral neutralization, or PCR (1,10,13,24, 25, 26, 27). The FAMA test (fluorescent antibody to membrane antigen) is useful for diagnosis of skin scrapings or biopsies. Cytopathologic results are similar to those of HSV, with Cowdry-type intranuclear inclusions and syncytia.
loss in the developed world (23). VZV is fastidious and hard to recover from clinical samples. Human fibroblast lines are best for tissue culture recovery, and likelihood of recovery is increased by culturing vesicular fluid located around the eye or on the V-1 dermatome. Cytopathic effect appears between 5 and 28 days in culture. Because of this difficulty, VZV is usually identified by immunofluorescence, viral neutralization, or PCR (1,10,13,24, 25, 26, 27). The FAMA test (fluorescent antibody to membrane antigen) is useful for diagnosis of skin scrapings or biopsies. Cytopathologic results are similar to those of HSV, with Cowdry-type intranuclear inclusions and syncytia.
CMV is a lymphotrophic herpesvirus and contains the largest genome in this family. It infects up to 2.5% of all newborns and 50% of the world’s adult population and is a frequent cause of congenital defects and an opportunistic virus in immunocompromised patients (2,10,28). Although CMV rarely causes problems in the immunocompetent host (other than CMV mononucleosis), with the dramatic rise in the number of immunosuppressed patients due to HIV infection and iatrogenic immunosuppression in the past two decades, CMV retinitis has become a major cause of visual morbidity and vision loss (29, 30, 31). All layers of the retina are damaged, and both intranuclear and intracytoplasmic inclusions are present.
Unlike other viruses, where either DNA or RNA are carried into the host cell, research indicates that CMV carries mRNA in the virion and that this is the source of infection. The virus replicates only in human cells, but these include a wide variety such as epithelial cells, fibroblasts, and macrophages. It establishes both persistent and latent infection, often asymptomatic, in mononuclear leukocytes, bone marrow, kidney, and heart. The virus has been isolated from virtually all forms of body fluid, from tears to blood or breast milk. Transmission is by the congenital, oral, and sexual routes, blood transfusion, and tissue transplantation. Diagnosis is made by a wide variety of tests such as isolation, immunoassays, and PCR testing, as discussed earlier. Serology is useful only for primary infection because of the persistence of the infection. Histopathology or cytology may show the diagnostic “owl’s eye” inclusion body (32,33).
EBV is the epitome of a lymphotrophic virus. It infects the B lymphocyte (B cell) and some epithelial cells of the nasopharynx, and causes or is causally associated with a variety of diseases such as infectious mononucleosis, endemic Burkitt’s lymphoma, Hodgkin’s disease, and nasopharyngeal carcinoma. This limited tissue tropism is a result of the limited cellular expression of its receptor. By the third decade of life, 90% of adults are positive for EBV antibody (2). Contagion rate is low, with most cases of mononucleosis contracted by intimate contact with an asymptomatic person shedding EBV from the oropharyngeal epithelium. Active infection may persist for years in mucosa-associated lymphoid tissue (34,35). The virus has never been isolated from tear film.
EBV encodes more than 70 proteins, various groups of which are expressed depending on type of infection: replicative, latent, or immortalization. During replicative infection, transcription and translation of the ZEBRA transcriptional activator protein activates the early viral genes, leading ultimately to synthesis of viral DNA, capsid, and glycoproteins. The viral proteins produced during the replicative infection are serologically defined as early antigen, viral capsid antigen, and membrane antigen, all of which are diagnostically useful in serologic testing (3,4). If the infection is a nonpermissive B-cell disease (latent), the cells contain only a few circular, plasmid-like EBV genomes that replicate only when the cell replicates. Immediate early genes are expressed along with Epstein-Barr nuclear antigens, which are also diagnostically useful. EBV is unique among herpesviruses in its ability to immortalize B cells.
The Epstein-Barr nuclear antigens and certain membrane proteins are DNA-binding proteins essential for establishing and maintaining infection and immortalization. They activate cell growth and prevent cell apoptosis (programmed cell death). The EBV genome is an episome in latent state and continuously expresses EBV nuclear antigen. B-cell production results in an IgM antibody to the Paul-Bunnell antigen, the heterophile antibody, another protein of diagnostic use. T cells usually control B-cell proliferation but, if control is lost, lymphoma will develop (2,3).
HHV8 DNA is most likely transmitted sexually. It has been detected in 95% of biopsy specimens from Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease through PCR testing and Southern blot analysis (36). It has also been found in the peripheral monocytes of HIV-positive patients. The virus may infect B cells, null cells, vascular endothelial cells, and perivascular spindle cells (37). HHV8 encodes proteins homologous to human proteins that enhance the growth and prevent apoptosis of infected and surrounding cells. This in turn promotes growth of polyclonal Kaposi’s sarcoma cells in immunosuppressed patients. It is also found in the B cells of approximately 10% of immunocompetent people.
Adenoviruses
Family: Adenoviridae
Genus: Mastadenovirus (mammals), human adenovirus
There are approximately 100 serotypes of adenovirus, of which at least 47 infect humans. They are subtyped on the basis of DNA homology and hemagglutination patterns into six subgroups, A through F. There is 90% genomic homology within an adenoviral type and 20% homology between adenoviral types. Adenoviruses are nonenveloped, icosadeltahedron viruses (70 to 90 nm in diameter) with linear double-stranded DNA at the core. The capsid is made of 240 capsomeres that consist of hexons and 12 pentons. Each penton has a fiber that contains the viral
attachment proteins, which have viral specific antigens and can also act as hemagglutinins (38,39) (Fig. 14-2).
attachment proteins, which have viral specific antigens and can also act as hemagglutinins (38,39) (Fig. 14-2).
The viruses are highly epitheliotropic with a small host range. The diseases caused include respiratory and gastrointestinal infections, hepatitis, cystitis, and keratoconjunctivitis. These viruses are endemic and cause illness year round. Ocular disease is transmitted primarily through direct contact with infected material such as saliva fomites or contaminated towels, or through bathing in a contaminated swimming pool. Immunity is lifelong but type specific (4,38,39). Replication of a single virus takes approximately 36 hours and produces 10,000 progeny. The virus binds to the host cell surface first by the penton fiber attaching to a glycoprotein, and then enters the cell by receptor-mediated endocytosis in a coated vesicle. The virus lyses the endosome and the DNA is delivered to the cell nucleus. Genomic transcription occurs on both viral DNA strands and in both directions at different times in the cycle. Early transcription produces 20 nonstructural proteins that stimulate cell growth and viral DNA replication. After early replication ends there is concurrent production of viral DNA in the nucleus mediated by DNA polymerase, and late transcription of structural proteins. Capsid proteins are produced in the cytoplasm and the capsids are then transported to the nucleus for DNA insertion. The process is inefficient, with only 1 infectious unit produced per 11 to 2300. The virus is released on cell lysis (4,38,39).
Diagnosis may be made in the office or in the laboratory. Adenoclone (Cambridge Bioscience, Worcester, MA) is an EIA that detects adenoviral antigen on conjunctival swabs (40). It is simple, rapid (1 hour), and inexpensive, and is 81% sensitive and 100% specific if taken within 1 week of disease onset. Laboratory diagnosis can be made by direct detection in clinical specimens or by viral isolation on human embryonic kidney or human HeLa or HEp-2 cell lines, with subsequent identification using fluorescent antibody or ELISA tests. Serotyping may be done after isolation from tissue culture. Other tests include serology of acute and convalescent sera, especially hemagglutination inhibition. A fourfold rise in antibody titer is diagnostic of adenoviral infection (4,10).
Poxviruses
Family: Poxviridae
Subfamily: Chordopoxvirinae
Genus: Orthopoxvirus: vaccinia, smallpox (variola)
Genus: Molluscipoxvirus: molluscum contagiosum virus, orf
The members of this virus family are the largest and most complex of all animal viruses, being almost visible on light microscopy and measuring 230 × 300 nm in a morphologically complex, bricklike shape with (3,41). The natural hosts for poxvirus that can infect humans are vertebrates in the bovine family. With the exception of smallpox and molluscum contagiosum, human infection occurs through accidental or occupational exposure. Smallpox and molluscum are spread from human to human. Smallpox is very contagious and spread primarily by the respiratory route and to some extent by contact with dried, but living virus, on cloth or other materials. Dissemination in the host then occurs by lymphatic and cell-associated viremia. Molluscum is spread by direct physical contact or by contact with infected material such as a towel. The lesions do not spread extensively.
Poxvirus replication is unique among the DNA agents in that the entire cycle occurs in the host cell cytoplasm. The virion therefore carries several enzymes, including DNA-dependent RNA polymerase, to allow this cytoplasmic replication. After fusion into the cytoplasm through host phagocytic vacuoles, viral RNA polymerase transcribes approximately half of the viral genome into early mRNA. The subsequent protein products include an enzyme that completes uncoating, plus TK and other enzymes (42,43). After uncoating, DNA synthesis begins and host macromolecular synthesis is stopped during this 1.5- to 6-hour period. The areas of DNA construction are seen as inclusion bodies in the cytoplasm. The viral DNA genome is then transcribed, but only late mRNAs are translated to structural proteins. Assembly of DNA and capsid then occurs and is completed by de novo synthesis of viral membrane by the poxviruses themselves. This viral membrane confers infectivity. Nucleocapsids that bud through taking just the host membrane are not infectious (43) (Fig. 14-2).
Smallpox and vaccinia have only recently become infectious agents of concern again, the former having been eliminated from the world by 1980. Now, however, it is a potential bioterrorism agent. Along with its preventative, vaccinia vaccination, both of these agents are of importance in ocular disease such as cellulitis, conjunctivitis, and acute or chronic keratitis or iritis. Useful diagnostic tests are not routine serologic tests but ELISA, radioimmunoassay, or monoclonal antibody assays (2,10,41).
Human Immunodeficiency Virus
Family: Retroviridae
Subfamily: Lentivirinae
Genus: Lentivirus (HIV types 1 and 2)
Genus: HTLV-BLV (human T-lymphotropic virus types 1 and 2-bovine leukemia virus)
The Lentivirinae, as the name implies, are characterized by very long incubation and latency periods (2,44). These medium-sized viruses (80 to 130 nm) have a positive-sense, single-stranded RNA genome, the viral enzymes protease, reverse transcriptase, and integrase, and a viral capsid.
There is broad genetic variability because of a very high level of virus turnover and low fidelity of reverse transcripts (2,10,45). These viruses share tissue tropism for the hematopoietic and neurologic systems and the ability to cause immunosuppression: acquired immunodeficiency syndrome (AIDS) in the case of HIV. More than 30 million adults and 10 million children are infected worldwide. In the United States and Europe, HIV-1 is the most common etiologic agent of this severe and often fatal illness. HIV-2 is more common in western Africa. It has a 55% homology with HIV-1 but causes a more benign clinical course with a much lower viral load in infected persons compared with HIV-1. Furthermore, HIV-2 is less frequently transmitted either sexually or vertically through a family.
There is broad genetic variability because of a very high level of virus turnover and low fidelity of reverse transcripts (2,10,45). These viruses share tissue tropism for the hematopoietic and neurologic systems and the ability to cause immunosuppression: acquired immunodeficiency syndrome (AIDS) in the case of HIV. More than 30 million adults and 10 million children are infected worldwide. In the United States and Europe, HIV-1 is the most common etiologic agent of this severe and often fatal illness. HIV-2 is more common in western Africa. It has a 55% homology with HIV-1 but causes a more benign clinical course with a much lower viral load in infected persons compared with HIV-1. Furthermore, HIV-2 is less frequently transmitted either sexually or vertically through a family.
Both HIV type 1 and 2 genomes have three genes common to all retroviruses: gag, pol, and env. The cell-derived viral membrane contains transmembrane glycoproteins gp41 and gp120, crucial to viral adherence. Gp120 binds to CD4 receptors on human T lymphocytes, and gp41 is necessary for fusion. After penetration, the virus uncoats and transcription of viral RNA proceeds through reverse transcriptase. HIV DNA then inserts itself into the host cell genome. The virus may remain latent, producing little or no mRNA, or be active, with virus-encoded RNA and proteins produced. Viral assembly takes place in the cytoplasm and viral exit is by budding through the host membrane (2,4,45,46) (Fig. 14-2).
Transmission is largely through unprotected sex and intravenous (IV) drug use with contaminated needles. Others at risk are recipients of blood transfusions and newborns of infected mothers. There is currently no preventive vaccine. The virus infects a variety of tissues, including blood, brain, lymph nodes, marrow, skin, and bowel (47). In the eye, it is permissive for anterior and posterior segment opportunistic infections and may itself cause disease of various structures of the anterior segment.
Diagnosis may be made by viral isolation (which takes up to 5 weeks), or the much faster techniques of serology or nucleic acid detection (2,10,48). The reliability of diagnosis is affected by the length of time between transmission and the time of testing. Serology, such as the enzyme immunoassay for IgG antibodies to HIV using viral lysate or recombinant proteins and the antigen, is of great value as a rapid and sensitive screening test. ELISA is also very sensitive for HIV-1, HIV-2, and HTLV, and is used for blood donation screening. Because of its lower specificity, however, the results are always confirmed by Western blot, immunoprecipitation, or indirect immunofluorescence. Western blot detects antibody to the specific HIV antigens p24, gp41, or gp 120. Antibody may be detected within 2 to 6 weeks of transmission but may take longer. If results are still equivocal, as in an early infection, detection of viral RNA by PCR is a reliable method of diagnosis (1,10,49). These immunologic/molecular virologic tests produce results in hours to a day or two.
The risk for development of AIDS in asymptomatic HIV-positive patients may be predicted with reasonable reliability by determining the number CD4+ helper T lymphocytes and viral load in the peripheral blood (CD4+ helper lymphocytes <200 cells/mL, viral burden >107 plasma HIV RNA copies/mL). This test, coupled with testing plasma HIV RNA levels, is a useful guideline for eligibility for antiretroviral or other antiviral therapy (e.g., for CMV retinitis), predicting disease progression, and monitoring disease control.
Human Papillomaviruses
Family: Papovaviridae
Genus: Papillomavirus: human papillomavirus (HPV)
The papillomaviruses are common throughout nature, highly epitheliotropic, genus-specific, and found most often among higher vertebrates (2,4,50). In humans, the viruses have been associated with warts, dysplasias, and carcinomas of the genital tract and conjunctiva (51,52). Mucosal areas are infected by HPVs 6, 11, 16, and 18, whereas skin sites are infected by HPVs 1 through 4. The viruses cannot be grown in tissue culture. Differentiation of the 70 different HPV types and understanding the reproductive cycle has been aided by molecular and immunologic virologic technology (1,3,10).
HPV is a nonenveloped, relatively small, 45- to 55-nm icosahedral structure containing linear double-stranded DNA complexed with histones of cellular origin (4,50,52). Genomic structure among the various papilloma viruses is similar and the major capsid protein represents 80% of the total viral protein (Fig. 14-2). The viral genome is divided into three areas: early, late, and regulatory. In replication, the early regions are necessary for transformation and the late region controls transcription and replication (51).
Diagnosis is based on morphology under electron microscopy and viral nucleic acid detection. Immunoperoxidase testing may be performed but is unreliable in intraepithelial neoplasia and invasive carcinoma because of absence of HPV antigens. Southern blot technique and in situ hybridization are useful, but limitations in our knowledge of the role of HPV in malignancy make the significance of detecting HPV DNA in lesions unclear (10,50,52).
Paramyxoviruses
Family: Paramyxoviridae
Genus: Paramyxovirus (mumps, parainfluenza, Newcastle disease)
Genus: Morbillivirus (measles virus)
Genus: Pneumovirus (respiratory syncytial virus)
The paramyxoviruses have similar morphology and protein components and a common capacity to induce syncytia and multinucleated giant cells (cell-cell fusion).
The major diseases caused by these agents are often common childhood infections that are now largely prevented through vaccination in the United States but remain a problem in underdeveloped nations (2,3). The viruses are large (156 to 300 nm) with a negative-sense, single-stranded RNA in a helical nucleocapsid surrounded by an environmentally labile membrane. Replication begins by viral hemagglutination protein attachment to the cell surface. A viral F transenvelope protein allows penetration by membrane fusion. Cells lacking the enzyme necessary to cleave the F protein are not permissive for viral infection. After penetration, viral RNA polymerase transcribes the negative-stranded RNA viral genome. Viral gene transcription and translation occur in the cytoplasm. The nucleocapsids assemble here as well and align along sites of membrane hemagglutination protein and F viral protein insertion. Exit is by budding with the acquisition of a host cell lipid envelope (2, 3, 4,10) (Fig. 14-2).
The major diseases caused by these agents are often common childhood infections that are now largely prevented through vaccination in the United States but remain a problem in underdeveloped nations (2,3). The viruses are large (156 to 300 nm) with a negative-sense, single-stranded RNA in a helical nucleocapsid surrounded by an environmentally labile membrane. Replication begins by viral hemagglutination protein attachment to the cell surface. A viral F transenvelope protein allows penetration by membrane fusion. Cells lacking the enzyme necessary to cleave the F protein are not permissive for viral infection. After penetration, viral RNA polymerase transcribes the negative-stranded RNA viral genome. Viral gene transcription and translation occur in the cytoplasm. The nucleocapsids assemble here as well and align along sites of membrane hemagglutination protein and F viral protein insertion. Exit is by budding with the acquisition of a host cell lipid envelope (2, 3, 4,10) (Fig. 14-2).
Both measles and mumps are highly contagious and transmitted by respiratory droplets (2,53, 54, 55). Measles has only one antigenic type. The incubation period after exposure is 9 to 11 days, with local replication in the respiratory tract preceding viremia, with seeding of leukocytes and the reticuloendothelial system. This is followed by a secondary viremia that seeds the skin (maculopapular rash), cornea, and conjunctiva. One third of infected patients do not manifest clinical disease but may transmit it. Diagnosis is based on clinical findings, but the virus may be grown in simian or human tissue culture, and shell-vial immunoassay and reverse transcription PCR are used for definitive tissue diagnosis such as in the late measles complication, subacute sclerosing panencephalitis (10). Acute and convalescent antibody titers are diagnostic if there is a fourfold rise.
Mumps has an incubation period of 7 to 25 days, averaging 8 days (2,53). Acquired by respiratory inhalation of virions, replication occurs in the upper respiratory tract, followed by a viremia and viral spread to other organ systems such as the parotid gland, testicles, ovaries, and brain (4,56). Ocular involvement may be widespread in both anterior and posterior segments. Diagnosis is usually clinical but fluorescent antibody stainings of conjunctival scrapings are also reliable. Other diagnostically useful tests include acute and convalescent sera, neutralization complement fixation and hemagglutination, and ELISA testing.
Newcastle disease virus is a helically symmetric, pleomorphic virus with a negative-stranded RNA at the core surrounded by a lipoprotein envelope containing hemagglutination and fusion glycoprotein fibers (2,10,53). Human infection is usually accidental from exposure to infected chickens. The incubation period is 1 to 2 days, followed by a conjunctivitis (57). Virus culture is possible but rarely used.
Togaviruses
Family: Togaviridae
Genus: Rubivirus: rubella virus (German measles)
Rubella virus is an enveloped, relatively small (50 to70 nm) icosahedral agent. Like measles, there is only one serotype (Fig. 14-2). Transmission is by inhalation of droplets from an infected patient. The illness is subclinical or mild, causing rash, fever, and lymphadenopathy, but its consequences for the unborn child of an infected woman can be devastating both systemically and ophthalmologically (2,58, 59, 60).
Diagnosis is based on serologic detection of anti-rubella antibodies or viral isolation (10). Viral shedding may remain positive up to 18 months after the acute illness. Because no cytopathic effect is produced in tissue culture such as simian or human kidney cells supporting viral replication, fluorescent antibody staining is used to shorten detection time to 48 to 96 hours rather than waiting for enterovirus interference testing. Detection of rubella IgM in infants is also diagnostic, whereas IgG is not because it crosses the placental barrier.
Coxsackieviruses
Family: Picornaviridae
Genus: Enterovirus: coxsackieviruses, human enteroviruses
The enteroviruses are in the same genus as the polioviruses. There are 67 serotypes in the enterovirus group. These are all very small viruses (30 nm) with a single strand of positive-sense RNA at the core and surrounded by a nonenveloped viral capsid (Fig. 14-2). These viruses may cause a wide variety of illnesses involving a wide variety of organ systems, including encephalitis, myocarditis, pleurodynia, neonatal sepsis, polio, peripheral neuritis, and acute hemorrhagic conjunctivitis (2,61). No serotype is associated with any one illness.
OCULAR ANTIVIRAL AGENTS: LABORATORY AND CLINICAL STUDIES*
Of the 10 ocular antiviral agents discussed in the following sections, the last 4 are essentially used only for CMV retinitis despite activity against herpetic, adenoviral, or vaccinia viral infection (23,62, 63, 64, 65). They are, however, occasionally used for the latter infections because of resistance to the first six drugs or because of ongoing studies into their potential use in ocular viral disease. Their review is brief and oriented to treatment of anterior segment disease.
Idoxuridine
Structure
Idoxuridine (5-iodo-2′-deoxyuridine; IDU; Herplex), a pyrimidine antimetabolite introduced commercially in 1962, was the first drug used to control human viral disease (66,67). Structurally, idoxuridine is a thymidine analog with the 5′-methyl group replaced by iodine. Because of the development of better and more convenient drugs, idoxuridine’s use and marketing have dropped dramatically in recent years.
Mechanism of Action
Idoxuridine is incorporated as a thymidine analog preferentially into viral DNA over cellular DNA because the former replicates more rapidly in infected cells (68,69). The drug is not selectively activated but is phosphorylated by both viral and cellular kinases. The idoxuridine triphosphate (IDU-TP) also inhibits the virus-specific DNA polymerase more than the host cell polymerases. Idoxuridine may interact specifically with deoxythymidine monophosphate synthetase, or it may be incorporated directly into DNA as idoxuridine monophosphate (70). Substitution of idoxuridine for thymidine in the DNA chain leads to abnormal transcription and translation, thus producing defective viral progeny. Its incorporation into some host DNA adds to the toxicity of the agent.
Although idoxuridine and its phosphorylated metabolites inhibit a variety of cellular enzymes, idoxuridine action as an antiherpetic agent may also be at a viral biosynthetic level before incorporation into the viral genome. Phosphorylation of idoxuridine to IDU-TP allows this compound to mimic deoxythymidine triphosphate, and subsequently IDU-TP may exert either an allosteric or a feedback inhibitory effect at the level of deoxythymidine kinase, deoxycytidine monophosphate deaminase, or cytidine diphosphate reductase, ultimately terminating viral replication (66,70).
The toxic effects of idoxuridine are magnified in tissues undergoing rapid cellular DNA synthesis (71). Clinical trials using the drug systemically for HSV encephalitis revealed that the drug was minimally effective and highly toxic (72). The drug is teratogenic and mutagenic, and potentially carcinogenic. For these reasons it is not used as a systemic agent.
In the eye, adverse reactions to topical idoxuridine include contact dermatitis, punctate epithelial keratopathy, follicular conjunctivitis, lacrimal punctal occlusion, and lid margin keratinization (73). Controlled studies on the effects of antivirals on corneal epithelial and stromal wounds indicate that idoxuridine is the most toxic of the commonly used topical antiviral drugs, interfering primarily with stromal healing and causing toxic epithelial changes (74).
Penetration and Pharmacokinetics
Pavan-Langston and coworkers demonstrated that in rabbits treated with 0.5% idoxuridine ointment, the drug did not penetrate the intact cornea (75). In human ocular penetration studies, idoxuridine, as the parent nucleoside, did not enter the aqueous humor. Instead, a metabolic breakdown product of idoxuridine, uracil, was detected in the aqueous. Only in patients in whom the corneal epithelium was damaged did idoxuridine penetrate the corneal barrier. When administered systemically (IV, intramuscularly, or subcutaneously), the drug is dehalogenated and rapidly metabolized; 50% to 75% is excreted within 4 to 5 hours after systemic administration in animals (76,77).
Efficacy
Idoxuridine is used topically for HSV keratoconjunctivitis. It is, however, effective in vaccinia keratitis (78, 79, 80). In vitro studies demonstrated that idoxuridine did not affect the adsorption of HSV in tissue culture cell monolayers, nor did it alter the infectivity of extracellular herpesvirus. idoxuridine did stop viral replication after it had begun and resulted in at least a 2-log-unit diminution in viral titer 24 hours posttreatment (81). The antiviral efficacy of idoxuridine was substantiated by numerous controlled clinical trials (82,83). Healing rates vary from approximately 75% in complicated cases to 90% in straightforward dendritic or geographic keratitis. When 0.1% idoxuridine was instilled into the eyes of patients with dendritic lesions, relief was noted within 12 to 24 hours after initiation of therapy. By 72 hours, notable healing of the epithelium occurred. Idoxuridine is highly effective in treating infectious corneal epithelial herpetic disease but is ineffective in HSV-induced iritis, stromal keratitis, and other forms of HSV intraocular infection (82,84). It is also less effective than trifluridine and vidarabine if topical steroids are being used concomitantly (85,86).
Idoxuridine use essentially stopped with the advent of newer, more convenient drugs; recommended therapy when used is 0.1% every 1 to 2 hours by day and 0.5% idoxuridine ointment at bedtime for 14 days.
Resistance
HSV resistance to idoxuridine is through modification of the TK gene. Clinically resistant virus strains isolated from patients or created by serial passage through idoxuridinecontaining medium all had the common finding of reduced TK activity, sometimes to as low as 5.6% of that of the wild type or the parental strain (87,88). This has significance in this time of the increasing emergence of resistant viral strains during treatment of immunosuppressed patients. Idoxuridine-resistant HSV showed cross-resistance to bromovinyldeoxyuridine and to acyclovir, intermediate
resistance to trifluridine, and full sensitivity to vidarabine and ganciclovir (88).
resistance to trifluridine, and full sensitivity to vidarabine and ganciclovir (88).
Trifluridine (Trifluorothymidine)
Structure
Trifluridine or trifluorothymidine [5-trifluoromethyl-2′-deoxyuridine, 2′-deoxy-5-(trifluoromethyl) uridine; TFT; F3T; Viroptic] is a fluorinated pyrimidine nucleoside. Structurally, it is an analog of the deoxyribonucleoside thymidine and is virtually identical to idoxuridine with the exception of three fluorine atoms attached to a methyl radical replacing the iodine (89). As a pyrimidine nucleoside analog, trifluridine inhibits DNA viruses, and because of its structure, it is incorporated rapidly into growing host cell types (e.g., bone marrow.)
Mechanism of Action
Although the specific mechanism of trifluridine inhibition of active herpesvirus production is not entirely certain, its antiviral activity appears similar to that of idoxuridine and results from the effect of the drug on viral DNA synthesis (89). Trifluridine can exert its inhibition at several stages in the viral biosynthetic pathway, starting from 2-deoxyuridine-5′-monophosphate to DNA synthesis. Trifluridine acts as a competitive inhibitor of thymidine (90). The drug is initially phosphorylated to trifluridine monophosphate, which is a potent inhibitor of thymidylate synthetase (91). After phosphorylation to the triphosphate form, the compound competitively inhibits incorporation of thymidine triphosphate into viral DNA.
Toxicity
The drug is cytotoxic, teratogenic, mutagenic, and potentially carcinogenic, and is sufficiently toxic that it is not used systemically (92,93). In vivo treatment of normal rabbit corneas with trifluridine caused no adverse effects or evidence of corneal toxicity (94). However, when rabbit corneas with standardized epithelial defects were treated with either 1% or 0.1% trifluridine drops eight times a day for 8 days, pathologic changes in the regenerating epithelium were observed. Stromal wound healing appears to be affected by trifluridine therapy (tensile strength was significantly reduced 12 days after wounding and treatment with 1% trifluridine) (95).
Penetration Studies and Pharmacokinetics
The biphasic solubility profile of trifluridine enhances the transport of the intact, active drug across the cornea. The mechanism of penetration (as for idoxuridine and vidarabine) appears to be by nonfacilitated diffusion as demonstrated by linear penetration kinetics through excised, perfused rabbit corneas and the lack of demonstrable saturation kinetics (96). In a controlled study, trifluridine penetrated the corneal epithelium faster than idoxuridine and vidarabine. The presence or absence of an intact epithelial cell layer did not significantly alter trifluridine distribution. However, the concentration of trifluridine in the aqueous was increased in debrided or damaged corneas, and the rate of penetration was doubled (97). Trifluridine (1%, four times a day) administered to both infected rabbit eyes and patients with a history of recurrent herpetic keratitis penetrated the corneal stroma and achieved therapeutic levels (5.3 to 18 μg/mL) (96, 97, 98).
The parent compound is hydrolyzed to 5-carboxy-2′-deoxyuridine within 3 to 5 hours at 37°C and pH 7.2 (97,99,100). This metabolite has little or no antiviral activity. In vivo, trifluridine is hydrolyzed to 5-carboxyuracil or 5-carboxyuridine with the loss of the inorganic fluoride. The rate of elimination of trifluridine correlates with the rate of trifluridine metabolism. Monophosphate, diphosphate, and triphosphate trifluridine metabolites are found in body tissues The short serum half-life (12 to 30 minutes) and toxicity of trifluridine prevent the use of this drug in systemic herpetic infections.
Efficacy
Trifluridine interferes not only with the replication of HSV-1 and HSV-2 but has an effect on vaccinia and certain adenoviruses (101). Trifluridine (0.2 to 1.7 μg/mL) inhibits the cytopathic effects of HSV-1 by 50% in plaque reduction assay (102). Plaque formation was reduced by over 98% when HSV-1 grown into Vero cells was treated with 17 μg/mL trifluridine (103). Trifluridine activity in vitro is comparable to that of idoxuridine, and trifluridine is considerably more active on a weight-for-weight basis than is vidarabine. As observed for both idoxuridine and vidarabine, the strain of HSV-1 appears to be of major importance in determining the relative antiviral efficacy. Trifluridine was shown to inhibit five strains of HSV-1 within a narrow range; however, the susceptibility of five HSV-2 strains was variable, with two strains being insensitive at the maximum nontoxic concentration (104).
Trifluridine was more potent on a weight-for-weight basis than idoxuridine in the treatment of HSV-1 herpetic keratitis in rabbits. When trifluridine and idoxuridine were compared with respect to their ability to eradicate virus from the preocular tear film, no virus was recovered on days 2 and 4 of the 7-day treatment with trifluridine. However, HSV-1 was present in idoxuridine treated eyes throughout the treatment regimen (82,105). Two days after discontinuation of therapy, rebound virus shedding had occurred in both the trifluridine and idoxuridine groups, with virus titers higher than those observed in
control, placebo-treated animals. These results indicate that a critical time period exists in an acute herpetic infection during which continued presence of the antiviral is necessary to control rebound virus shedding, even though infectious virus cannot be detected in the tear film. Clinical studies comparing topical 1% trifluridine with 0.1% idoxuridine drops, 3.0% vidarabine, or acyclovir ointments have shown that, overall, the latter two drugs and trifluridine have efficacy rates of 90% to 95% regardless of whether steroids are in use (85,106, 107, 108, 109, 110). The idoxuridine overall efficacy rate was approximately 76%, dragged down by apparent decreased efficacy in the face of steroid use in some patients, or perhaps by its having been in clinical use so long that certain organisms had become resistant or patients had become allergic to the agent. Although trifluridine had a slight edge over all other drugs in the face of concomitant steroid therapy, no statistical difference could be shown.
control, placebo-treated animals. These results indicate that a critical time period exists in an acute herpetic infection during which continued presence of the antiviral is necessary to control rebound virus shedding, even though infectious virus cannot be detected in the tear film. Clinical studies comparing topical 1% trifluridine with 0.1% idoxuridine drops, 3.0% vidarabine, or acyclovir ointments have shown that, overall, the latter two drugs and trifluridine have efficacy rates of 90% to 95% regardless of whether steroids are in use (85,106, 107, 108, 109, 110). The idoxuridine overall efficacy rate was approximately 76%, dragged down by apparent decreased efficacy in the face of steroid use in some patients, or perhaps by its having been in clinical use so long that certain organisms had become resistant or patients had become allergic to the agent. Although trifluridine had a slight edge over all other drugs in the face of concomitant steroid therapy, no statistical difference could be shown.
Vidarabine
Because of the availability of trifluridine and oral antivirals, vidarabine (9-B-D-arabinofuranosyladenine, vidarabine, ara-A, Vira-A) is no longer commercially available in the United States. For special conditions such as ocular vaccinia, however, it is available through compounding pharmacists.
Structure
9-B-D-Arabinofuranosyladenine is a purine arabinosyl nucleoside structurally similar to deoxyguanosine (112).
Mechanism of Action
Like all current antivirals, vidarabine is virostatic. Its biologic activity is due in part to intracellular phosphorylation, first to the monophosphate form (ara-A-MP) and subsequently to diphosphate and triphosphate forms (ara-A-DP, ara-A-TP) by cellular enzymes. Vidarabine inhibits viral DNA synthesis by several mechanisms. Direct incorporation into DNA as ara-A-MP results in DNA chain termination. The triphosphorylated form, ara-A-TP, inhibits terminal deoxynucleotidyl transferase and DNA polymerase (δ and β). Ara-A-DP and ara-A-TP also inhibit the activity of ribonucleotide reductase enzymes (113, 114, 115).
Toxicity
After systemic or topical administration, vidarabine is converted to a hypoxanthine arabinoside derivative (ara-HX); this metabolite has only 20% of the antiviral potency of the parent compound (116). The toxicity of vidarabine, administered systemically or topically, is negligible over a wide range of pharmacologic dosages. A major problem in systemic administration of vidarabine is that the compound’s relative insolubility requires the use of large volumes of fluid, which can tax cardiovascular and renal homeostatic mechanisms. The drug is also teratogenic and mutagenic, and has carcinogenic potential (117). As a result, vidarabine is used systemically only as an alternative to acyclovir or ganciclovir in drug-resistant, life-threatening HSV or VZV infections.
Vidarabine administered topically to rabbits did not impair wound healing as measured by planimetry and histopathologic examination. However, stromal wound strength of corneas treated topically with vidarabine or idoxuridine was less than that of wounds treated with placebo ointment (74).
Penetration and Pharmacokinetics
Ocular penetration studies have shown that topically applied vidarabine crosses the corneal barrier in minor amounts in corneas with intact epithelium. The deaminated semiactive metabolite ara-HX was found in the aqueous in significant amounts after topical vidarabine treatment (75,97). Application of 3% vidarabine in a watermiscible cream produced corneal levels of 20 μg/mL, whereas the same concentration in petrolatum ointment produced levels of only 4.5 μg/mL.
After IV administration, vidarabine is rapidly deaminated by adenosine deaminase to the less effective ara-HX form or other noneffective metabolites. Plasma levels of ara-HX are directly related to the rate of vidarabine infusion. Vidarabine’s plasma half-life is approximately 3.5 hours, and the metabolite ara-HX is well distributed in tissues. Ara-HX readily crosses the blood-brain barrier and attains concentrations within 35% of plasma levels in the cerebrospinal fluid (CSF) (116,118). To enhance the antiviral efficacy of vidarabine therapy, potent inhibitors of adenosine deaminase, which stop the conversion of vidarabine to ara-HX, have been used. These inhibitors have enhanced the virucidal activity (by 20-fold) and the cytotoxic activity of vidarabine in vitro and in vivo (119,120).
Efficacy
Vidarabine has a broad antiviral spectrum that includes many DNA viruses (HSV-1, HSV-2, vaccinia, VZV, CMV, and pseudorabies virus); it has little or no effect against RNA viruses (80,84,121, 122, 123). Plaque reduction assays
using several different herpesvirus strains indicated a high degree of strain variability. As is the case with idoxuridine, the cell type, virus passage, and virus strain all significantly affect the antiviral activity of vidarabine. Vidarabine is highly effective against idoxuridine-resistant HSV-1 and HSV-2 in vitro (80).
using several different herpesvirus strains indicated a high degree of strain variability. As is the case with idoxuridine, the cell type, virus passage, and virus strain all significantly affect the antiviral activity of vidarabine. Vidarabine is highly effective against idoxuridine-resistant HSV-1 and HSV-2 in vitro (80).
In the eye, topical vidarabine 3% ointment has been found effective in herpetic keratitis and keratouveitis in both animal models and in humans (86,107,124). In one study of 69 patients with external ocular herpetic keratitis, vidarabine proved to be equal to idoxuridine in reducing tear film viral titers and in promoting corneal reepithelialization (125). Vidarabine was significantly less toxic than idoxuridine and caused healing of herpetic lesions clinically resistant to idoxuridine. Other studies in patients with extensive dendrogeographic ulcerations substantiate these findings (124). Fewer treatment failures occurred with vidarabine (9.5%) compared with idoxuridine (18.8%) in a study of more than 300 patients with ocular herpes (86). Several studies referenced in this section and the section on trifluridine indicate that there is no statistically significant difference between topical vidarabine, trifluridine, and acyclovir therapy of HSV keratitis (HSK) (107,109,124).
Systemic vidarabine has also been effective in therapy of varicella and in herpes zoster ophthalmicus (HZO), although it is now in distant fourth place behind valacyclovir, famciclovir, and acyclovir in this regard. Whitley et al. reported that vidarabine therapy (10 mg/kg/day for 5 days) of varicella in immunocompromised patients reduced fever duration, lesion count, and systemic morbidity significantly compared with placebo (122). A Collaborative Antiviral Study Group report on vidarabine therapy of zoster infection (including HZO) in immunocompromised patients noted that the same therapy used for varicella, mentioned previously, resulted in more rapid cessation of viral shedding and increased healing rate of lesions (126). Postherpetic neuralgia (PHN) could not be evaluated. Its role in TK-negative, acyclovir-resistant HSV mutants continues to make this drug of interest (127, 128, 129).
Resistance
Resistance to vidarabine appears related to viral DNA polymerase rather than to TK. Mutant, vidarabine-resistant viruses with changes in the DNA polymerase gene have been isolated (130,131). This resistance was mapped to a 0.8-kb region in the pol gene and makes viral DNA polymerase less susceptible to ara-A-TP (130,132). Although other gene products may be affected, most HSV mutations have been found to code for the carboxyl-terminal portion of the viral DNA polymerase (133). Such DNA polymerase mutation is of clinical significance because mutant HSV is resistant not only to vidarabine but to the other common agents that use this antiviral mechanism. Resistance to vidarabine is uncommon and is discussed with reference to its use in HSV and VZV infections resistant to other antivirals (idoxuridine, acyclovir).
Acyclovir
Structure
Acyclovir [9-(2-hydroxyethoxymethyl)guanine; acyclovir, acycloguanosine (Zovirax)] is a synthetic compound that was designed to mimic substrates for model enzyme systems. Acyclovir is an analog of guanosine or deoxyguanosine in which the 2′- and 3′-carbon atoms are missing. Acyclovir was the first compound engineered to have selective in vitro and in vivo antiviral activity (102,134,135).
Mechanism of Action
The major modes of action of acyclovir are viral DNA chain termination and rapid inactivation of viral DNA polymerase. Acyclovir is phosphorylated (activated) to acyclovir monophosphate (ACV-MP) specifically by herpesvirus-encoded TK, thereby largely bypassing activation in any but infected cells (102,134). This markedly reduces toxicity and increases specificity. ACV-MP is further phosphorylated to ACV diphosphate (ACV-DP) and ACV triphosphate (ACV-TP) by viral and cellular enzymes. ACV-TP competes for deoxyguanosine triphosphate, the natural substrate for virus-specific DNA polymerase, and is incorporated irreversibly as ACV-MP onto the growing viral DNA chains, thus causing chain termination (136,137). Termination occurs because acyclovir lacks the 3′ hydroxyl group needed to react with incoming nucleotides. DNA polymerase then binds irreversibly to the acyclovir-terminated chain and the entire enzyme complex is metabolically inactive. Little to no inactivation of cellular polymerase occurs (102,136, 137, 138). Elion and coworkers have determined that ACV-TP inhibits HSV DNA polymerase (DNA nucleotidyl transferase) 10 to 30 times more efficiently than cellular DNA polymerase, another factor in reducing toxicity (133,134). Approximately a 3000-fold concentration increase in acyclovir (above that which is virucidal) is needed to inhibit host cell growth.
Toxicity
Because acyclovir is activated specifically by herpesviruscoded TK, it was anticipated that the drug would be relatively nontoxic. After corneal application of 3% acyclovir ointment, no toxic corneal effects were noted, and the quality and rate of reepithelialization and stromal wound strength of the cornea were not impaired (74). Clinical study did note rare diffuse punctate keratitis that cleared after discontinuation of acyclovir therapy, but this apparent toxicity was thought to be a function of the drug vehicle (139).
The only important metabolite of acyclovir is 9-carboxymethoxy-methyl guanine, an inactive compound that accounts for up to 14% of acyclovir dosage administered to humans (140). Renal clearance of acyclovir ranges from 75% to 80% of the total body clearance and is substantially greater than the clearance of creatinine, indicating that glomerular filtration and tubular secretion mechanisms (possibly the organic acid secretory system) are involved (141). The drug may rarely cause renal failure when given in high doses (>5 mg/kg) IV if there is renal insufficiency or dehydration. Hydration of all patients should be monitored and patients with renal insufficiency should have reduced doses and prolongation of the intervals between dosing to minimize this risk. Rare central nervous system (CNS) toxicity, delirium tremens, and coma have been reported (142). Other common side effects are gastrointestinal distress and headache. There is no evidence that acyclovir is a carcinogen or teratogen, and, at therapeutic doses, there is no effect on the hematopoietic or immune systems (143).
Penetration and Pharmacokinetics
Acyclovir can be administered safely and effectively by IV or subconjunctival injection, orally, or topically. Its adsorption after oral administration is variable and appears to be species dependent (140,143, 144, 145). Animal studies have indicated that after oral administration in mice, 50% of the acyclovir is adsorbed and peak plasma concentrations occur 2 to 4 hours after ingestion (141).
In humans, oral adsorption of acyclovir is incomplete, with bioavailability of the drug ranging between 15% and 30% of the dosage (146). Peak plasma concentrations occur 1.5 to 2.5 hours after administration, and steady-state plasma concentrations after multiple oral doses of 2.5, 5, 10 and 15 mg/kg administered every 8 hours were 6.7, 9.7, 20, and 20.6 μg/mL, respectively. These values are similar to the peak plasma concentrations observed after equivalent single oral doses, and it was concluded that acyclovir does not accumulate in plasma after repetitive dosing. Oral dosing of 200 mg every 4 hours reaches steady-state levels ranging from 1.4 to 4.0 μM, which is inhibitory for HSV-1 and HSV-2, but doses of 800 mg orally (PO) five times daily are needed to yield peak and trough serum levels, respectively, of 6.9 μM and 3.5 μM to inhibit most strains of VZV (147, 148, 149).
Studies by Hung et al. and Collum et al. on the concentrations of acyclovir in the tear film and aqueous in patients on 400 mg PO five times daily showed levels of 3.28 μM (0.96 to 8.79 μM) and 3.26 μM (1.10 to 5.39 μM), respectively, 4 hours after the last oral dose (147,150). The mean ED50 (effective dose reducing viral plaque count in tissue culture by 50%) for HSV-1 ranges from 0.15 to 0.18 μM to 0.1 to 1.6 μM, which indicated that the tear and aqueous levels achieved were well in excess of that which should be needed to eliminate the virus (68,148,150, 151, 152). This is not always the case. Stromal HSV particles may persist in the face of prolonged, therapeutic doses of oral acyclovir (153). Drug resistance may have played a role in this finding. Compared with HSV, the inhibitory doses for VZV are much higher, at 3 to 4 μM, resulting in the need for fourfold higher drug dosing, as noted earlier, and less leeway in terms of resistance (152).
After topical instillation of 3% acyclovir ointment in the inferior cul de sac of 25 eyes (every 5 hours for four to six doses before cataract extraction), the mean acyclovir concentration in the aqueous humor was 1.7 μg/mL, which falls within the therapeutic range (152,154). Cutaneous adsorption of acyclovir may occur through damaged skin, but systemic adsorption is limited.
Total-body distribution of IV 14C-acyclovir has been studied in several animal species (144). Acyclovir rapidly entered all tissues after administration in both mice and rats. In humans with normal kidney function the serum half-life is approximately 3 hours. IV dosing of 5 mg/kg three times daily resulted in serum ID50 levels (inhibitory dose reducing viral plaque count in tissue culture by 50%) well above those needed for HSV-1 and HSV-2 at all times, but a dosage of 10 mg/kg three times daily was necessary to avoid trough levels that fell below the ID50 of several VZV strains (144,148,154,155). Multidose IV therapy with 400 to 1200 mg/m2 every 8 hours resulted in acyclovir concentrations in kidney, lung, and brain that were 1000%, 131%, and 25% to 70% of plasma levels, respectively (156). CSF acyclovir levels were approximately 50% of corresponding plasma levels. The rapid penetration of acyclovir into the CSF has made this drug an important compound for treating focal and disseminated CNS herpetic infections. Further information is given in the section on valacyclovir, later.
Intravitreal levels in humans 2 hours after 13 mg/kg IV acyclovir were at inhibitory levels for HSV-1 and HSV-2, VZV, and EBV (147). Regular acyclovir dosing of 5 mg/kg three times daily yielded concentrations of 8.8 to 11.0 μM, well in excess of the inhibitory dose for HSV-1 and HSV-2, VZV, and EBV, as did 25 mg of subconjunctival acyclovir (157).
Clinical Efficacy
In vitro studies with acyclovir have demonstrated that this compound has a broad antiviral spectrum, including HSV-1, HSV-2, VZV, EBV, and, to a much lesser extent, CMV (135). It is not effective against vaccinia. Acyclovir is 160 times more potent than vidarabine and 10 times more potent than idoxuridine (158,159). Inhibitory doses for various viruses were noted previously.
Herpes Simplex Virus
Acyclovir (100 μg/mL) has been used in a drug-induced suppression model of HSV-1 infection in trigeminal
ganglion cells. In this system, it produces a suppressed HSV-1 infection that is functionally identical to HSV-1 latency in vivo. Acyclovir has also been shown to suppress the reactivation of latent virus in explanted trigeminal ganglia (160). Latency was not eradicated, however, as demonstrated by an increase in viral titer after acyclovir removal. The rabbit eye model of HSV-1 infection has been used extensively to assess the in vivo efficacy of acyclovir (161,162).
ganglion cells. In this system, it produces a suppressed HSV-1 infection that is functionally identical to HSV-1 latency in vivo. Acyclovir has also been shown to suppress the reactivation of latent virus in explanted trigeminal ganglia (160). Latency was not eradicated, however, as demonstrated by an increase in viral titer after acyclovir removal. The rabbit eye model of HSV-1 infection has been used extensively to assess the in vivo efficacy of acyclovir (161,162).
It is not within the purview of this chapter to cover in fine detail the multiple clinical uses of acyclovir, except to note that this important drug is indicated or has been effective in the following conditions: (a) primary genital HSV (PO or IV), (b) recurrent genital HSV in immunocompetent patients (PO), (c) mucocutaneous HSV in immunocompromised patients (PO or IV), (d) HSV encephalitis (IV), (e) neonatal HSV (IV), (f) varicella in immunocompetent (PO) or immunocompromised patients (IV then PO), (g) herpes zoster in immunocompetent (PO) or immunocompromised patients (IV the PO), and (h) possibly EBV infections (PO) (107,127, 163, 164, 165).
Multiple clinical studies on infectious HSV epithelial keratitis comparing topical 3% acyclovir with 3% vidarabine ointment, 0.1% idoxuridine drops, or 1.0% trifluridine drops in recommended doses revealed no statistically significant difference among the four drugs, although there was a trend suggesting that trifluridine, acyclovir, and possibly vidarabine were superior to idoxuridine if concomitant steroids were given (23,109,124,139,149, 164,166, 167, 168, 169). Acyclovir ophthalmic ointment is not commercially available in the United States although it is marketed in other countries.
Oral acyclovir 400 mg five times daily is equivalent to topical acyclovir in treating epithelial keratitis, with 90% of patients healing their ulcers in a mean of 5 days (147,170, 171, 172). Two hundred milligrams PO five times daily healed 18 of 19 patients with combined HSV epithelial and stromal keratitis in 5 to 21 days (171). The therapeutic pediatric dosage is 20 to 40 mg/kg/day for 7 to 14 days as a pediatric elixir (200 mg/tsp) (170).
Between 1994 and 2001, eight multicenter studies on the efficacy of oral acyclovir or topical steroids, or both, on various forms of ocular HSV were reported from the Herpetic Eye Disease Study (HEDS) (173, 174, 175, 176, 177, 178, 179, 180). The results may be briefly summarized as follows:
After resolution of any form of ocular HSV, 1 year of acyclovir 400 mg twice daily significantly reduced recurrence of herpetic disease during that time and without rebound up to 6 months after acyclovir was stopped.
There was no statistically significant benefit of acyclovir 400 mg five times daily for 10 weeks in treating active HSV stromal keratitis already being treated with steroids and trifluridine.
Steroids were significantly better than placebo in resolving active stromal keratitis, and postponing steroids slowed resolution but showed no difference in outcome by 6 months.
Treatment of iritis with acyclovir 400 mg five times daily for 10 weeks in patients already on steroids and trifluridine may possibly have some beneficial effect.
A 3-week course of acyclovir 400 mg five times daily for patients with epithelial keratitis already on trifluridine did not alter the subsequent incidence of stromal keratitis or iritis.
Acyclovir 400 mg twice daily for 1 year significantly reduced the recurrence of HSV stromal or epithelial keratitis, with greatest benefit in the stromal group.
Previous stromal, but not epithelial, keratitis markedly increased the risk of recurrent similar disease in the future.
Psychological stress, sun exposure, contact lens wear, or systemic illness could not be shown to be triggers for HSV reactivation.
The number of past episodes of either epithelial or stromal keratitis was strongly associated with the likelihood of a recurrence.
Long-term oral acyclovir significantly lowers recurrence of either form of HSK, but is more effective in preventing recurrence in stromal than in epithelial disease.
It should be noted that prophylactic use of oral antivirals is legitimate but defined as “off label” use.
In a study of 105 patients with HSK, Wu and Chen reported an even more positive prophylactic effect in preventing recurrent epithelial HSV in patients who had not undergone keratoplasty than did the HEDS (174,181). Using low-dose acyclovir at 300 mg/day for 1 year, they found statistically significant differences between treated and control groups: 5 recurrences of epithelial and 1 case of stromal keratitis in the acyclovir group, and 14 cases of epithelial and 4 cases of stromal keratitis in the untreated control group. The epithelial data conform with those reported by Simon and Pavan-Langston (182).
There are additional indications for oral acyclovir in patients with herpetic keratitis. These include use as an adjunct to topical antivirals in patients with atopic disease or in immunosuppressed patients, especially in patients with AIDS (23,127,183, 184, 185, 186). Oral or IV therapy is determined on the basis of severity of immunosuppression in AIDS (CD4+ T-helper lymphocytes <200 cells/mL, viral burden >105 to 107 plasma HIV RNA copies/mL) and degree of illness (187). Dosage in atopy of 400 mg PO three times to four daily for 2 to 3 weeks is usually quite effective. Another indication is use in those patients who are unable or unwilling to take topical antiviral agents for epithelial keratitis, such as those with crippling arthritis, children or uncooperative adults, those whose occupation makes topical agents difficult to use, and those with ocular
toxic medicamentosa from local antivirals. Adult dosage is 400 mg PO three to five times daily, and for children, 20 to 40 mg/kg in divided doses for 7 to 10 days. Prophylaxis of HSV epithelial recurrences with oral acyclovir in patients post-HSV keratoplasty is effective and indicated. It is discussed in further detail later in the section on Surgical Factors and Management (182,188, 189, 190).
toxic medicamentosa from local antivirals. Adult dosage is 400 mg PO three to five times daily, and for children, 20 to 40 mg/kg in divided doses for 7 to 10 days. Prophylaxis of HSV epithelial recurrences with oral acyclovir in patients post-HSV keratoplasty is effective and indicated. It is discussed in further detail later in the section on Surgical Factors and Management (182,188, 189, 190).
Varicella Zoster Virus/Herpes Zoster Ophthalmicus
The role of acyclovir in HZO is now well established (23,149,151,164,184,191, 192, 193, 194, 195, 196, 197). In studies of topical acyclovir, McGill and coworkers reported that 5% acyclovir ointment five times daily was highly effective in resolving zoster epithelial keratitis and in significantly reducing the incidence of recurrent disease compared with topical steroids (198,199). In addition, the combination of topical steroid and acyclovir was found to be less effective than acyclovir alone, and there was no difference among groups in resolution of stromal keratitis, iritis, scleritis, or secondary glaucoma. In contrast, Marsh and Cooper reported an overall trend for more rapid resolution of inflammation in patients with HZO treated with the acyclovir-steroid combination, compared with the progressive ocular inflammation noted in patients treated with topical acyclovir alone (200). No patient received systemic acyclovir or corticosteroids. The study concluded that topical steroids were useful in the management of the inflammatory aspects of HZO and that there was no clear benefit of topical acyclovir ophthalmic ointment when used alone.
Conversely, Zaal et al. have reported that at 3 months after onset of HZO, patients who received 3% topical acyclovir had longer durations of periocular lesions and significantly more visual loss compared with the group receiving oral acyclovir, and that chronic disease developed in all patients put on combined topical acyclovir and dexamethasone drops (201). Acyclovir ointment is not available in the United States, but is in use in Europe and South America. The aforementioned studies and the data on systemic acyclovir all suggest that the systemic route is the one to use in treating VZV.
In immunocompetent patients, acyclovir 800 mg PO five times daily for 7 to 14 days (average, 10 days) results in more rapid resolution of viral shedding, acute neuralgia, and new skin lesions, and a reduced incidence of pseudodendritiform keratopathy, stromal keratitis, and iritis. There was variable effect on PHN, depending on the study, and no effect on corneal hypesthesia or neurotrophic ulceration. With the exception of PHN, acyclovir compares favorably with famciclovir and valacyclovir (see later). In the presence of immunosuppression, where HZO tends to be more severe and slow to respond to therapy, initial treatment with IV acyclovir is indicated (1500 mg/m2/d in three divided doses = 10 to 15 mg/kg every 8 hours) for 7 to 10 days, followed by acyclovir 800 mg PO five times daily for 6 to 14 weeks. This dosage is similar to that used in acute retinal necrosis (202,203). Because patients with HZO are at increased risk for complications, several studies have been conducted demonstrating that use of oral acyclovir in ophthalmic zoster has a high therapeutic efficacy. Compared with placebo, acyclovir therapy, 800 mg PO five times daily for 7 to 10 days, induces a prompt resolution of skin rash, cessation of pain, more rapid healing, reduced duration of viral shedding, and reduced duration of new lesion formation. There is also a significant reduction in the incidence and severity of acute dendritiform keratopathy, scleritis, episcleritis, iritis, the incidence (but not the severity if it occurred) of corneal stromal immune keratitis, and the incidence of late-onset ocular inflammatory disease (e.g., episcleritis, scleritis, iritis) (197,204, 205, 206, 207). Effect on PHN was variable, with some reports showing no efficacy, and others notable decrease in severity and incidence (197,202,206, 207, 208, 209). Two studies noted that higher dosages of oral acyclovir, 800 mg given orally five times daily, for 7 to 10 days had beneficial effects similar to those noted by others, but also reduced the prevalence of localized zoster-associated neurologic symptoms such as paresthesia, dysesthesia, and hyperesthesia. With the exception of PHN, acyclovir compares favorably with famciclovir and valacyclovir (see later) (207,210). The current U.S. Food and Drug Administration (FDA)-approved adult dose for HZO is 800 mg PO five times daily for 7 days.
The FDA-approved dosing recommendations for acyclovir are as follows: (a) genital HSV, 200 mg PO five times daily for 10 days (first episode) or for 5 days (recurrence); (b) HSV prophylaxis, 400 mg PO twice daily; (c) acute zoster, 800 mg PO five times daily for 7 to 10 days; (d) IV, 5 to 10 mg/kg every 8 hours, each dose over 1 hour; (e) safety and efficacy of acyclovir not established in children younger than 2 years of age; (f) varicella, 20 mg/kg PO four times daily for 5 days; and (g) use adult dose if patient weighs 40 kg. The American Association of Pediatrics does not recommend routine use of acyclovir for varicella, but consider use if patient is older than 12 years of age, has chronic cutaneous or pulmonary disease, or is on long-term steroid or salicylate treatment. Off-label use of acyclovir in HIV-positive adults is as follows: (a) genital HSV, 400 mg PO three time daily for 7 to 10 days, and for 5 to 10 days for recurrence; and (b) genital HSV prophylaxis, 400 to 800 mg PO two to three times daily (211,212). Ocular doses are usually adapted from genital HSV study doses if there has been no specific ocular study done.
Resistance
An important consideration in the use of TK-activated antivirals is the development of resistant HSV strains. In vitro studies have shown that acyclovir resistance can be
manifested by (a) a lack of viral TK (TK−), (b) altered substrate specificity of viral TK such that it phosphorylates thymidine but not acyclovir, and (c) a viral DNA polymerase gene mutation to alter the enzyme such that it is not inhibited by ACV-TP. By far the most common mechanism of resistance is TK− mutation (213,214).
manifested by (a) a lack of viral TK (TK−), (b) altered substrate specificity of viral TK such that it phosphorylates thymidine but not acyclovir, and (c) a viral DNA polymerase gene mutation to alter the enzyme such that it is not inhibited by ACV-TP. By far the most common mechanism of resistance is TK− mutation (213,214).
Acyclovir-resistant HSV and VZV mutants are rarely seen in immunocompetent patients, but resistant strains are being encountered ever more frequently in immunodeficient patients (215, 216, 217). The AIDS epidemic has accelerated this because of large numbers of patients on long-term acyclovir therapy. Despite the TK− status of most mutants, severe mucocutaneous disease may result. Vidarabine and foscarnet are reasonable alternative drugs because they do not require TK for activation (131,218,219). Sonkin et al. have reported isolation of an acyclovir-sensitive HSV with normal TK activity from a patient who had undergone keratoplasty on postoperative day 22 (220). Five and 7 days later, as the clinical course deteriorated, acyclovir-resistant HSV with deficient TK activity was isolated. Despite sensitivity to foscarnet, the graft eventually failed.
Valacyclovir
Structure
Valacyclovir (VCV; Valtrex) is the L-valine ester of acyclovir. This prodrug was synthesized to enhance greatly the oral absorption of acyclovir from the gastrointestinal tract. It undergoes an almost complete first-pass conversion to acyclovir and L-valine through enzymatic hydrolysis. The bioavailability of acyclovir is enhanced three to five times by this prodrug and is not altered by the simultaneous administration of food.
After enzymatic conversion to acyclovir, a 1-g dose of valacyclovir gives peak plasma concentrations of 5 to 6 μg/mL of acyclovir, a therapeutic dose for both HSV and VZV (221, 222, 223, 224). One gram of valacyclovir orally four times daily gives a concentration-time curve of acyclovir similar to that with the IV administration of 5 mg/kg of body weight of acyclovir every 8 hours (222).
Mechanism of Action
After enzymatic conversion to acyclovir, the mechanism of action of valacyclovir is identical to that discussed previously for acyclovir.
Toxicity
In general, toxicity is negligible and patient tolerance is excellent (225). Carcinogenicity, tumorigenicity, fertility, and mutagenicity studies on valacyclovir were negative (222). Although acyclovir does cross the placenta, it was found that the rate of birth defects in infants of women on acyclovir during pregnancy was the same as the general population. The registry of patients studied was too small to make definitive conclusions about the safety of the drug in the fetus.
Valacyclovir does, however, have a potential significant toxicity in immunosuppressed patients. Thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome has been reported in bone marrow or renal transplant recipients or HIV-positive patients who were taking high doses of valacyclovir for prolonged periods. This drug therefore is not indicated in immunocompromised patients. There have been no such adverse reports in immunocompetent patients (183,222).
Penetration and Pharmacokinetics
The pharmacodynamics of valacyclovir are similar to those of acyclovir (221,222,224,226). Acyclovir is widely distributed through all body organs and fluids, including the brain, CSF, vaginal mucosa, uterus, seminal and herpetic vesicular fluids, liver, and kidneys. Although there are no studies on the intraocular penetration of valacyclovir, it seems safe to assume that drug titers are similar to or higher than those discussed previously for acyclovir. Acyclovir concentrations after oral valacyclovir are approximately 50% of plasma levels. After multiple doses of 1 g of valacyclovir, these levels are 5 to 5.5 g/mL. Eighty to 89% of drug is eliminated by the kidneys as acyclovir. Mean renal clearance of acyclovir exceeds creatinine clearance, indicating that renal tubular secretion is actively involved in elimination of drug, which is 99% acyclovir.
Clinical Efficacy
Valacyclovir’s antiviral activity, in descending order of in vitro susceptibility, is HSV-2, HSV-1, VZV, EBV, HHV6, HHV8, HHV7, and CMV (224,227, 228, 229, 230). It is not effective against vaccinia. The primary FDA-approved clinical uses of valacyclovir are for treatment of herpes zoster and herpes simplex (222,224,231,232).
Clinical studies comparing valacyclovir 1 g PO three times daily with acyclovir 800 mg PO five times daily in 1141 immunocompetent patients with herpes zoster (35 with HZO) indicated that the drugs were equivalent in ability to accelerate dermal healing and reduce the duration of virus shedding, but that valacyclovir was significantly better in acute pain resolution and reduced duration of PHN up to 1 year of follow-up (196,224). There was no difference if drugs were given 7 or 14 days. Further, studies on PHN revealed that the medium time to pain resolution was 38 days with valacyclovir and 51 days with acyclovir (p < .03). Other studies support the good efficacy of valacyclovir in herpes zoster, particularly if started within 72 hours of rash onset (22,233, 234, 235, 236).
The only ocular study compared acyclovir with valacyclovir in 121 immunocompetent patients with acute HZO, and reported an incidence of keratitis, uveitis, and episcleritis that was similar in both groups (237). Neither group had any incidence of neurotrophic keratitis or scleritis, and acute pain was noted in approximately two thirds of each group. It was concluded that valacyclovir was a valid alternative to acyclovir in treatment of HZO, but, like famciclovir (see later), was superior in acute and long-term pain inhibition and in patient compliance, with only three-times-daily dosing.
Clinical studies in immunocompetent patients using the well-tolerated valacyclovir 1000 mg PO three times daily indicate equivalent efficacy with acyclovir 400 mg PO five times daily in acute zoster.
Currently used dosages have been adopted from genital HSV studies (231,232). These reports indicate that for acute HSV infection, valacyclovir 1000 mg PO every day for 10 days is as effective as acyclovir at 200 mg PO five times daily for the same time period. For recurrent genital HSV, valacyclovir therapy is 500 mg PO two times daily for 3 days, and for suppression of recurrent episodes of HSV, 1 g PO every day (226,231, 232,238). The same valacyclovir dose has been used for long-term prophylaxis up to one year with no adverse side effects in immunocompetent patients.
The FDA approved dosages of valacyclovir are (a) for primary genital HSV, 1 g PO two times daily for 10 days; (b) for genital HSV prophylaxis in HIV-positive patients, 500 mg PO twice daily; (c) for varicella in young adults, 500 mg PO three times daily for 5 days; (d) and for orolabial HSV in immunocompetent patients, 2 g PO q12h × 2 treated as soon as possible (controversial use). Off-label use is for varicella in adolescents at 500 mg PO three times daily for 5 days (212,239). Ocular use is also off label, with doses as used in the above clinical studies. However, in some severely immunocompromised HIV-positive patients, thrombocytopenic purpura/hemolytic uremic-syndrome developed, with a few deaths (22). As a result, this drug is not FDA approved for use in immunocompromised patients.
Resistance
The mechanism of resistance of herpesviruses, most commonly VZV, to valacyclovir is similar to that for acyclovir—that is, the vast majority are TK deficient (226). Although VZV resistance emerging from valacyclovir treatment of immunocompetent patients is very rare, the incidence of acyclovir-resistant VZV in immunosuppressed patients is increasing. It is most common in HIV-positive individuals, but occasionally is seen in organ transplant recipients (213, 214, 215). Alternative therapy in these cases would be IV foscarnet (202,242).
Famciclovir
Structure
Famciclovir (Famvir) is the orally bioavailable diacetyl ester of 6-deoxy-penciclovir. Penciclovir itself is an acyclic guanine derivative similar to acyclovir in structure, mechanism of action, and antiviral activity.
Mechanism of Action
Penciclovir and acyclovir differ qualitatively in rates of phosphorylation, concentration of triphosphate derivatives, stability, and affinity for viral DNA polymerase (243, 244, 245). The intracellular activity of penciclovir is very long even when extracellular titers are low—for example, 10 to 20 hours and 9 hours in HSV- and VZV-infected cells, respectively, compared with less than 1 hour for acyclovir (ACV-TP) in similarly infected cells (246). Further, penciclovir triphosphate (PCV-TP), a nonobligate DNA chain terminator, is more effective than ACV-TP, an obligate chain terminator, in inhibiting HSV DNA polymerase-mediated DNA chain elongation (243,247).
Toxicity
Penciclovir’s preferential phosphorylation in herpesvirusinfected cells far exceeds that of acyclovir, but the minimal phosphorylation of penciclovir in uninfected cells and the low activity of PCV-TP against cellular DNA polymerases explains penciclovir’s and its derivative, famciclovir’s, lack of toxicity in cell culture and in clinical studies (244,245). There was no significant difference in adverse experiences such as headaches, nausea, and diarrhea between patients with zoster and genital herpes infections receiving oral famciclovir or placebo up to 18 weeks in masked studies. Laboratory abnormalities and results of hematologic analyses, clinical chemistries, semen analyses, and urinalyses were similar in both famciclovir and placebo groups (248,249).
Penetration and Pharmacokinetics
Although penciclovir is very poorly absorbed from the gastrointestinal tract, famciclovir has an absorption rate of 77% after oral administration. The metabolite, penciclovir, is not further broken down but is eliminated unchanged in the urine, with a half-life of approximately 2 hours after IV administration (244,245,250). As noted in the section on Mechanism of Action, intracellular PCV-TP activity persists despite low extracellular titers.
Efficacy
Famciclovir’s efficacy is similar to that of acyclovir. In vitro activity includes HSV-1, HSV-2, VZV, and EBV. It is not effective against vaccinia. Penciclovir’s in vitro activity against VZV equals that of acyclovir, with a mean EC50 in tissue culture of 3 to 4 μg/mL (244,245,250). In experimental HSK in rabbits, oral famciclovir twice daily at doses of 60 to 500 mg/kg body weight, regardless of dose, resulted in significant reduction in keratitis and HSV-1 genomes in the trigeminal ganglion, as well as improved survival. These data suggest that oral famciclovir may reduce the morbidity of HSV in clinical situations (231,251, 252, 253, 254, 255).
Clinical studies in several hundred immunocompetent patients with nonophthalmic zoster infection have been quite successful. Famciclovir, 500 mg PO three times daily, was compared with placebo or with acyclovir 800 mg five times daily for 7 days. Results were clearly superior to placebo and similar to acyclovir both in stopping viral shedding and accelerating dermal healing. Perhaps more important, famciclovir had a significantly greater effect on reduction of PHN compared with placebo or acyclovir. With an incidence of just over 50% in all groups, the median time to resolution of the neuralgia was 55 and 62 days in the famciclovir groups, compared with 128 days in the placebo group. These are also more favorable data on PHN than those seen in acyclovir-treated patients (253,256). A study comparing valacyclovir with famciclovir showed the drugs to be comparable in nonocular VZV infection in terms of cutaneous healing and pain resolution (235). This further affirms the superiority of both famciclovir and valacyclovir over acyclovir in acute zoster and, more important, PHN.
In HZO, a randomized, double-masked study on 454 patients comparing famciclovir 500 mg PO three times daily with acyclovir 800 mg PO five times daily revealed that both drugs were equally effective in all parameters of ocular disease (252). The effects on neuralgia were not reported.
The drug is FDA-approved for treatment of herpes zoster infection at dosages of 500 mg TID/day PO for 7 days. Dosage for recurrent genital herpes simplex is 125 mg PO twice daily for 5 days, and for herpes prophylaxis, 250 mg PO twice daily for a year or more. For acute orolabial HSV infection, the dose is 500 mg PO twice daily for 7 days (212,239). Ocular use is also off label, with doses as used in the aforementioned clinical studies.
Resistance
There have not yet been cases of reported resistance.
Ganciclovir
Structure
Ganciclovir [9-(1,3-dihydroxy-2-propoxy)methylguanine; DHPG; GCV; Cytovene, Cymevene] was the first of the five currently FDA-approved agents for treatment of CMV retinitis in immunosuppressed patients. It is also active, however, against HSV, VZV, and EBV. This nucleoside analog of deoxyguanosine is similar in structure to acyclovir, with the addition of a terminal hydroxymethyl group in the acyclic sugar (257). The active form is gancli-clovir-5′-triphosphate.
Mechanism of Action
Like acyclovir, ganciclovir activation in HSV and VZV infection begins with its monophosphorylation by virus-specific TK. Because ganciclovir is a far better substrate for viral TK, nearly 10-fold more ganciclovir triphosphate than ACV-TP is formed in HSV-1-infected cells (68,257,258). The triphosphate, in turn, selectively inhibits viral DNA polymerase.
Toxicity
Ganciclovir is effectively not metabolized by the body and its clearance depends on the kidneys. Although plasma levels and half-life increase with reduced renal function, the drug is not nephrotoxic. Toxicity is primarily hematopoietic, with neutropenia and thrombocytopenia, usually reversible, occurring in approximately 40% and 20% of patients, respectively, and often obviating use or limiting dosage of other myelosuppressive agents such as trimethoprim-sulfamethoxazole, amphotericin B, and zidovudine, but it may be used with other anti-HIV dideoxynucleotides (259, 260, 261). Half of each group sustains dose-limiting hematologic suppression; neutropenia is seen more frequently in patients with AIDS and thrombocytopenia in patients immunosuppressed due to other causes. Other, less frequent side effects include nausea, neurotoxicity, hepatic dysfunction, fever, and local rash or phlebitis (ganciclovir = pH 11). Ganciclovir is also carcinogenic and teratogenic, and induces azoospermia (127).
Penetration and Pharmacokinetics
Efficacy
Although ganciclovir is as active as acyclovir against HSV-1, HSV-2, VZV, and EBV, given its toxic nature, it is used only in life- or vision-threatening CMV infections. In addition to its FDA-approved uses, ganciclovir has notable potential as a topical anti-HSV agent. Clinical trials have shown that 0.15% ganciclovir gel or 3% acyclovir ointment three to five times per day over 1 week was therapeutically
equivalent, but that ganciclovir had superior local tolerance (264).
equivalent, but that ganciclovir had superior local tolerance (264).
Resistance
Ganciclovir-resistant CMV strains have been isolated in up to 27% of patients with retinitis receiving treatment for 9 months or longer (265). The mutation giving resistance to ganciclovir is in either the UL97 or UL54 gene. Genotypically resistant virus developed increasing phenotypic resistance with time and continued ganciclovir therapy. Change in therapy may allow a shift in virus population and the CMV genotype identified. Cidofovir, foscarnet, and vidarabine are alternatives in the face of ganciclovir resistance.
Cidofovir
Structure
Cidofovir [(S)-9-(3-hydroxy-2-phosphonylmethoxypropyl) cytosine; HPMPC; CDV; Vistide; Forvade] is an acyclic nucleotide analog derived from phosphonoformic acid. It is currently FDA approved in combination with probenecid for treatment of CMV retinitis in HIV-positive patients (266,267). Of equal interest, however, is the finding that this drug may be effective against several viruses of ocular importance, including HSV, vaccinia, and smallpox (268, 269, 270, 271, 272).
Mechanism of Action
The drug does not require activation by viral-encoded TK, and phosphorylation of cidofovir is independent of virus infection. The mechanism of action is through cidofovir diphosphate, which inhibits viral DNA polymerase at concentrations 50-fold lower than the level that inhibits cellular polymerases (268). The decreased rate of viral DNA synthesis is due to incorporation of cidofovir into the growing viral DNA chain (273,274). The drugs resist degradation by esterases and persist intracellularly up to 65 hours. This may allow drug-carrying cells to resist replication for several days when challenged by virus after therapy has been discontinued.
Toxicity
Cidofovir is carcinogenic in rats and potentially so in humans. At 65 times the recommended dose, it causes chromosomal alterations in red blood cells in vitro. It inhibits spermatogenesis and is embryotoxic in laboratory animals (273, 274, 275). Pediatric use should be with caution because of the carcinogenic potential and reproductive toxicity.
The major dose-limiting toxicity of cidofovir is nephrotoxicity in 50% of patients on the maintenance dose of 5 mg/kg body weight every other week. There is an early increase in serum creatinine and proteinuria, and later glucosuria when proximal tubular damage occurs. Neutropenia occurs in 20% of patients on maintenance. Interestingly, ocular hypotony was noted in several patients on maintenance (273).
Penetration and Pharmacokinetics
Pharmacokinetic studies in vitro show slow elimination, with a half-life of 22 hours, which supports use of intermittent dosing to achieve efficacy while minimizing toxicity (273,274). Protein binding is minimal (<6%) and the time to peak serum concentration is at the end of the infusion. Peak serum concentration with a 1-hour infusion of cidofovir 5 mg/kg body weight with probenecid is approximately 19 μg/mL. Approximately 70% of cidofovir is recovered unchanged in human urine within 24 hours.
Efficacy
Cidofovir is effective against many herpesviruses, including HSV-1, HSV-2, VZV, EBV, and ganciclovir-resistant andsensitive CMV, as well as against vaccinia virus and several adenoviruses (266,267,273,275, 276, 277). Comparison of topical 1% and 0.5% cidofovir with trifluridine and acyclovir in the rabbit HSV-1 keratitis model showed that both concentrations of cidofovir were significantly better than either of the other two drugs. (232, 233) Snoeck et al. have reported in vitro studies on vaccinia that of both phosphonate derivatives, cidofovir (HPMPC) proved to be the most therapeutically effective and most promising agent for both vaccinia and smallpox (271).
Cidofovir is of interest as a potential treatment for adenovirus. Romanowski et al. have reported in vitro efficacy against adenovirus types 1, 5, 8, and 19. In the rabbit model, 0.5% cidofovir twice daily for 7 days showed significant efficacy against adenovirus types 1, 5, and 6, all important strains in epidemic forms of the ocular disease (278,279). Antiviral prophylaxis against adenovirus-5 infection with twice-daily dosing of 0.5% or 1% drops was also highly effective at blocking viral replication (280).
Resistance
In experimental adenoviral ocular infection, treatment with 0.5% cidofovir twice daily for 7 days of known cidofovir-resistant strains showed no therapeutic effect compared with the parental adenovirus-5 strain in the rabbit model (276). In CMV retinitis, clinical resistance to intravitreal cidofovir was found in 5% of patients and was associated with prior oral ganciclovir or IV cidofovir. Resistance-associated mutations were found in the UL97 and
polymerase genes. Resistant CMV disease can result from a local infection independent of a systemic site of CMV infection (281).
polymerase genes. Resistant CMV disease can result from a local infection independent of a systemic site of CMV infection (281).
Foscarnet
Structure
Foscarnet (phosphonoformic acid; PFA; Foscavir) is a pyrophosphate derivative of phosphonoacetic acid. It differs from all previously discussed antivirals in that it is not a nucleoside analog (282,283). It is FDA approved for treatment of CMV retinitis in immunosuppressed patients, but is also of use in certain drug-resistant ocular viral infections, such as HSV and VZV.
Mechanism of Action
Antiviral action is through a selective and noncompetitive inhibition of viral-specific DNA polymerases and RNA polymerases (reverse transcriptases) at concentrations not affecting cellular polymerases (284). Because the reaction is reversible, the drug is virostatic, with viral replication restarting on removal of the agent. Because foscarnet does not require phosphorylation and directly affects the pyrophosphate binding site on viral polymerases, it is a therapeutically useful agent against resistant TK− herpesviruses. Because of its anti-viral reverse transcriptase effect, it has efficacy against HIV (68,128, 218,219,242,284,285).
Toxicity
Renal toxicity may occur in up to 45% of patients with AIDS with resulting azotemia and the need for constant monitoring of creatinine levels in all patients on the drug (282,286). The drug is excreted unmetabolized by renal tubular secretion and glomerular filtration. Toxicity may be due, in part, to foscarnet interference with renal phosphate secretion (287,288). This nephrotoxicity may be avoided or minimized in a number of cases by using intermittent foscarnet dosing rather than constant infusion and by maintaining excellent patient hydration. Elevated creatinine levels almost invariably return to normal (if due to drug) within 2 to 4 weeks of reducing or ceasing foscarnet administration. Electrolyte changes must also be monitored because the serum calcium may become quite high or low, a problem in itself, and one that makes concomitant use of other drugs that affect calcium balance, such as pentamidine, more precarious (289). Similarly, serum phosphorus and magnesium levels should be monitored.
Other toxic side effects include anemia in up to 50% of patients, hepatic enzyme elevation, CNS dysfunction, headaches, nausea and other gastrointestinal disturbance, mucous membrane erosions, and nephrogenic diabetes insipidus (282,288). Neutropenia and thrombocytopenia are not seen with foscarnet therapy.
Penetration and Pharmacokinetics
Oral absorption of foscarnet is very poor, resulting serum levels negligible, and gastrointestinal irritation common (282,288). The IV induction dose at the recommended regimen of 60 mg/kg over 1 to 2 hours three times daily results in peak plasma levels above the ID50 for most CMV (100 to 300 μM), HIV (132 μM), and HSV (100 μM) strains (128,282,288). CSF levels are approximately 43% of plasma levels (282). The half-life is 4 hours, and no drug is detectable in the serum by 24 hours, having been cleared by the kidneys unmetabolized. Unlike other antivirals, part of the foscarnet dose sequesters in the bone marrow for as long as several months (275). Over a 7-day period, up to 20% of the administered dose may be deposited in the marrow of patients with AIDS. This may be related to the calcium fluctuations seen in some patients.
Efficacy
Foscarnet is effective against all known human herpesviruses—CMV, HSV-1 and HSV-2, VZV, and EBV—and against the retrovirus, HIV. The primary therapeutic indication is the treatment of CMV retinitis in immunosuppressed patients. Other indications include any systemic form of life-threatening CMV infection, as well as acyclovir-resistant mucocutaneous HSV or VZV infection in immunocompromised patients (282). Doses of 60 mg/kg IV three times daily for 2 to 3 weeks and 90 to 120 mg/kg/day as maintenance are as effective as ganciclovir in CMV retinitis (282,290). Clinical response takes approximately 1 week. Because it does not need viral TK or other enzyme-dependent phosphorylation to be activated, the drug is also of use, and superior to vidarabine, in treatment of the acyclovir-resistant HSV and VZV infections most commonly seen in patients with AIDS (218,219,282).
Resistance
Recent reports of foscarnet-resistant HSV involved virus that was also acyclovir resistant (291). There was no relationship to the TK gene found to explain the acyclovir resistance. Four patients had undergone allogeneic stem cell transplantation. One was cured with valacyclovir and two with cidofovir, but the fourth patient died despite cidofovir treatment. A leukemic child with foscarnet-resistant HSV stomatitis responded well to a course of cidofovir (292).
HERPES SIMPLEX OCULAR DISEASE
HSV, the most ubiquitous communicable infectious virus in humans, is the causal agent of a wide variety of chronically recurring diseases. Ocular herpes simplex, caused primarily by type 1 (oral) but occasionally by type 2 (genital) HSV, is the leading infectious cause of corneal blindness in the United States, with approximately 500,000 cases reported annually (4,184,293,294). Recurrent herpes labialis and dermatitis, also usually caused by HSV-1, afflicts approximately one third of the world population; one half of these people experience more than one attack annually. Herpes genitalis, caused primarily by HSV-2, is the second most common venereal disease in this country, with approximately 100,000 cases reported annually (2,295). In neonates and children, the primary attack of HSV can be devastating. It may be associated with herpetic encephalitis and dissemination of infection, which have very high degrees of morbidity and mortality. Immunologically compromised patients, such as those with atopy, patients with leukemia, and organ transplant recipients, are also at high risk of severe systemic and ocular herpetic infections (2,184,185,294,296, 297, 298, 299, 300, 301, 302).
Incidence and Epidemiology
In contrast to the extensive data available on the epidemiology of extraocular HSV infection, little is known about the epidemiology of ocular infections. Nonetheless, information derived from nonocular herpetic studies is of use in understanding many aspects of the often visually debilitating ocular involvement.
The only natural reservoir of HSV is humans, with sources of infection being children with primary disease, adults with recurrent disease, and children and adults as healthy asymptomatic carriers. There is a very high incidence of herpetic neutralizing antibodies in children younger than 6 months of age, presumably from passive transfer through the placenta. This incidence then falls to approximately 20% in children from 6 months to 1 year of age, and then rises slowly to approximately 60% by age 15 to 25 years (2,294,303,304). In a sociologic study, Smith et al. demonstrated a decrease in the incidence of herpetic antibodies in a large population in England. They attributed this to a general improvement in the social environment, improved housing conditions, and better general hygiene (305).
Transmission of the virus appears to be primarily by direct contact. In the head area, this would be salivary droplets or direct oral contact leading to a primary infection, usually around the mouth. Occasionally, silent shedding with no apparent lesions may be the cause of viral spread to other patients (301). Although the primary illness is manifested clinically in only approximately 1% to 6% of patients infected with virus, the virus still has access by the oral route to the trigeminal ganglion, the ganglion that innervates the eye (305, 306, 307). The incubation period between contact and disease is from 3 to 9 days.
In an epidemiologic study on 141 patients with documented infectious epithelial HSK, Bell et al. found an overall predominance of male patients in the 80 patients older than 40 years of age (1.67:1.0; p < .03) (295). Of the 65 patients with more than one episode, 34% had a mean recurrence rate of one or more episodes annually and 68% had one or more recurrences every 2 years. The median interval between episodes was 1.5 years (range, 1.7 months to 20.8 years). No patient risk factors for frequent recurrence or for severity of disease (age of entering into study, age of first ocular episode, sex, or source of primary eye care) were noted. However, 46% of 57 recurrences in 27 patients occurred during the winter months of November through February, suggesting a possible role for upper respiratory tract infections in the pathogenesis of infectious ocular herpes.
Liesegang et al. found no seasonal trends in incidence, although rates increased with time (293). The overall prevalence of a history of ocular herpes was 149 cases per 100,000 population, and initial ocular episodes included incidences of 54% for blepharitis and conjunctivitis, 63% for epithelial keratitis, 6% for stromal keratitis, and 4% for uveitis. In a similar but prospective study by Shuster and coworkers of 119 percent with ocular herpes, the recurrence rates were somewhat lower, with 24% of patients having recurrences within 1 year and 33% within 2 years. This may reflect a difference in recurrence rates in the northeastern United States as opposed to the South and West. Shuster and colleagues also found no correlation between the rate of recurrence and either sex or age of the patient, but did note a general correlation between short intervals between past attacks and short intervals between future recurrences. This conforms with the HEDS data (177,308).
There is currently no vaccine for HSV, although killed, subunit, vaccinia hybrid, and DNA vaccines are under development to prevent spread of the virus and to treat infected patients. Glycoprotein D is used in several subunit vaccines, and live, defective mutant viruses lacking critical genes are being used in disabled infectious single-cycle vaccines. This latter vaccine produces noninfectious virions in the inoculated host (3).
Clinical Features
Ocular herpes may be classified into three general groups: congenital and neonatal, primary, and recurrent.
Congenital and Neonatal Ocular Herpes
HSV infection in the newborn may be acquired at one of three periods: intrauterine (5%), peripartum (10%), and
postpartum (85%). The first category occurs in 1/300,000 births, with ophthalmic of microphthalmia, retinal dysplasia, optic atrophy, and chorioretinitis (309). There are also often marked skin, vital organ, and neurologic lesions.
postpartum (85%). The first category occurs in 1/300,000 births, with ophthalmic of microphthalmia, retinal dysplasia, optic atrophy, and chorioretinitis (309). There are also often marked skin, vital organ, and neurologic lesions.
HSV infections in the latter two periods are further classified as skin, eyes, or mouth (SEM), with or without the other involvement seen in intrauterine infections. SEM presents at 10 to 12 days of age, whereas CNS disease usually appears at 16 to 19 days of age (309). Ocular herpes may include one or all of the following: conjunctivitis, epithelial keratitis, stromal immune reaction, cataracts, and necrotizing chorioretinitis (310,311). One third of neonates with HSV have CNS disease with or without SEM. In the era before the availability of IV vidarabine and acyclovir, only 18% of neonates had HSV disease limited to SEM. With the advent of acyclovir, this number has risen to 43% (166,309). Similarly, in the pre-systemic antiviral era, 38% of SEM neonates had developmental problems by 1 year of age. With vidarabine and acyclovir, these numbers dropped to 12% and 2%, respectively.
Hutchison et al. recovered HSV-1 from the eye of an infant, a dizygotic twin, born with active herpetic keratitis (312). The mother had no evidence of cervical herpes by culture or serial immune titers, delivery was by cesarean section, and there had been no premature rupture of the membranes. It was hypothesized (but it could not be proved) that the infection was acquired in utero by the transplacental, not the ascending, route. Periocular HSV infection also developed in the second twin a few days after birth. Both infants were treated with topical idoxuridine and, because of the chance of disseminated disease, systemic vidarabine, and both made a full recovery.
Although the vast majority of all ocular herpes infections are caused by HSV-1, because of the usual nature of acquisition, 80% of neonatal cases are caused by HSV-2 (298). Despite the unusual case reported by Hutchinson et al., neonatal disease is almost invariably secondary to direct exposure to an HSV-2-infected birth canal during the late prenatal period or during passage through an infected canal at birth itself (312). Inoda and colleagues reported a 3-year-old boy with stromal keratitis and iritis of unknown etiology. PCR testing of the aqueous fluid revealed HSV-2 as the causative pathogen. The patient recovered on acyclovir and steroid therapy (313). Multiple recurrences are far more common with genital and oral herpes than with ocular disease. Studies have shown an 89% recurrence rate for genital and a 42% recurrence rate for oral HSV infection over a 1-year period. In contrast, the ocular herpes recurrence rate was 40% over a 5-year period, which is fortunate, considering the visual consequences (184,314).
Infants’ “protective” transplacental antibodies are not sufficient to protect them completely from ocular disease, although they may escape life-threatening visceral involvement. In almost all reported cases, there has been an associated vesicular eruption of the skin, which is a most useful finding when the physician is faced with the need for a very rapid diagnosis. The clinical course of the ocular disease is similar to that encountered with primary or recurrent infections, depending on whether the infant has passively acquired transplacental antibody from the mother. Epithelial ulcers ultimately respond to antiviral therapy, but stromal involvement may clear or leave a nebulous scar. In the absence of superinfection, the skin lesions heal without scarring. An emergency pediatric or infectious disease consult is essential because IV acyclovir should be started urgently to minimize serious systemic complications of infection (315).
Primary Ocular Herpes
As noted earlier, most newborns are protected, in part, by maternal anti-herpetic antibodies transferred passively across the placenta in utero. This partial protection lasts for approximately 6 months, at which time the passively received antibodies have dwindled to negligible titers and the child becomes nonimmune and susceptible to a primary attack of HSV Fortunately, more than 94% of children do not suffer overt disease when they finally do contract the virus; those who do have overt disease usually manifest it about the mouth and not the eye. All those who are infected (60% by the age of 5 years) then become viral carriers, with the agent resting in latent state in the sensory (trigeminal ganglion) and autonomic nervous systems (304,316, 317, 318, 319, 320, 321, 322).
Although HSV has been isolated from the tear film of patients with no history of ocular herpes, studies on patients with a known history of herpetic keratitis show them to have no greater risk of asymptomatic shedding than does the normal population (323). In addition, tear antibodies to HSV-1 may be detected in the absence of detectable parotid saliva or serum antibodies. This suggests that the ocular surface may be an initial infection site for HSV in some healthy, clinically normal subjects and that there may be a preferential homing of committed B lymphocytes to the ocular mucosal surface (324). It is not yet determined what role, if any, these findings have in terms of latency and recurrent disease.
Primary ocular involvement may present as an acute follicular conjunctivitis or keratoconjunctivitis with nonsuppurative preauricular adenopathy and often with notable vesiculating periocular skin involvement (Fig. 14-3A). Pseudomembranes may be present in the fornices. In the absence of skin vesiculation, differentiation from adenoviral infection is aided by a careful search of the lid margins for signs of herpetic blistering.
Primary HSK is often atypical. Initially, there may be just a nonspecific, diffuse punctate keratitis that evolves into multiple scattered microdendritic figures. There may be wandering linear serpiginous ulcers across the entire corneal surface (184). The diffuse nature of this primary epithelial involvement is probably a function of the host’s nonimmune state, which allows more widespread ulceration. Primary
disease is, as a rule, confined to the epithelium in terms of clinical findings. Stromal involvement is not usually seen in this phase of the disease, presumably because the host is not immunologically programmed against the virus.
disease is, as a rule, confined to the epithelium in terms of clinical findings. Stromal involvement is not usually seen in this phase of the disease, presumably because the host is not immunologically programmed against the virus.
 FIGURE 14-3. A: Acute primary herpes simplex virus (HSV) blepharitis with confluent vesicles on hyperemic base. B: Recurrent HSV vesicles on lower lid showing much more localized lesions than in primary disease. |
Primary ocular herpes should not be confused with “first ocular occurrence.” The former is a first encounter with the virus, whereas the latter is the first eye involvement with HSV in a patient who has had a subclinical oral or nasal infection and is immune. First ocular occurrence is, therefore, similar to recurrent ocular herpes as described later.
Recurrent Ocular Herpes
Patients with recurrent herpes have both cellular and humoral immunity against the virus. The disease may present as any one or a combination of the following:
Blepharoconjunctivitis
Episcleritis, scleritis
Corneal:
Epithelial disease
Infectious ulceration
Trophic (metaherpetic) ulceration
Stromal disease
Viral necrotizing keratitis
Interstitial keratitis, immune rings, and limbal vasculitis
Disciform keratitis
 FIGURE 14-4. A, B: Two examples of herpes simplex virus corneal dendritic ulcers with terminal bulbs at the end of each branch. |
Endotheliitis
Iridocyclitis and trabeculitis
Blepharoconjunctivitis, Episcleritis, and Scleritis
Blepharoconjunctivitis is a much more localized infection than that seen in primary disease. Vesicles are localized rather than diffuse, starting as red papules that form clear vesicles, break, and scab over to heal without scarring (Fig. 14-3B). Virus is present for approximately 3 days in the lesions, although the lesions themselves take approximately 1 week to heal. Conjunctivitis is usually diffuse and watery. Occasionally, rose bengal or fluorescein staining reveals a conjunctival dendritic ulcer. Episcleritis and scleritis may occasionally occur and are slowly responsive to topical or oral nonsteroidal antiinflammatory agents (NSAIDs) such as ketorolac drops four times daily and ibuprofen. Topical steroids are effective in NSAID-unresponsive cases.
Epithelial Disease
Infectious Ulceration. HSV-induced eruptions of the corneal epithelium are characteristically thin, branching dendritic ulcers; wider, branching dendrogeographic ulcers; or map-shaped geographic lesions (Fig. 14-4). All are caused by live virus. Little inflammatory cell reaction-neutrophils but no lymphocytes-is seen in this form of ocular herpes, but many free viruses lie in intracellular and extracellular locations, particularly in the basal epithelium (325).
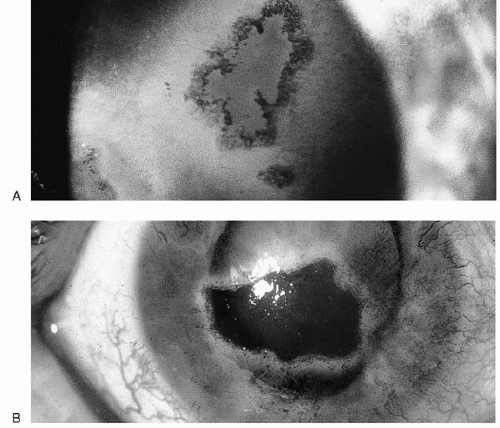 FIGURE 14-5. A, B: Two examples of scallop-edged infectious herpes simplex virus geographic corneal ulcers. These may be differentiated from trophic mechanical ulcers in that the latter have smoother, thicker edges. |
Signs and symptoms include tearing, irritation, photophobia, and often blurring of vision. Because the only presenting clinical finding may be a watery conjunctivitis, the patient should be asked about any trauma, previous corneal ulcers, inflammation inside the eye (iritis), nasal or oral cold sore, genital sores, recent use of topical or systemic steroid or immunosuppressive drugs, and immunologic deficiency states such as HIV positivity, malignancy, organ transplantation, or chronic eczema.
If corneal examination reveals dendritic or even geographic ulceration, the infectious agent is HSV until proved otherwise. In herpes, corneal sensation is reduced in approximately 70% of patients (326). It is almost never totally absent, as often seen in zoster keratitis. Marked stromal scarring as seen in more chronic forms of herpes results in a more marked decrease in sensitivity. Initially, the corneal lesions begin as a fine, transient, punctate keratitis that coalesces to form dendritic, dendrogeographic, or geographic ulcers (Fig. 14-5). In an eye that has had many recurrences or is receiving steroids without antiviral prophylaxis, the more subtle stages may be bypassed and the cornea may simply break down in rapidly forming geographic ulceration.
The actual incidence of infectious epithelial HSK in immunocompetent patients may be much higher than suspected clinically. Kodama et al., using immunofluorescent staining and corneal sensitivity testing, reported that of 48 eyes with diagnoses of nonherpetic conditions, 11 had positive reactions on immunofluorescent staining and 8 (73%) had decreased corneal sensitivity (326). The diagnosis in nine of these patients was recurrent erosion and in two, nonherpetic superficial punctate keratitis. Conversely, only 30% of eyes not suspected of having herpes keratitis and having negative findings on immunofluorescent staining still had decreased corneal sensitivity. Clinically, the nine “recurrent erosion” eyes that were not correctly diagnosed clinically as herpetic were described as having epithelial defects that were oval and central with hazy surrounding edges. Superficial stromal opacities were occasionally observed. It is likely, then, that had these eyes been correctly diagnosed as herpetic, they would not have been placed into the category of sterile trophic mechanical ulceration and therapy incorrectly directed toward lubrication and therapeutic lenses.
Contrary to common opinion, recurrent epithelial infections are not always due to the patient’s original HSV strain. Remeijer et al. have reported a study on 30 patients in whom sequential corneal HSV-1 isolates revealed that 63% were genotypically the same from recurrence to recurrence, whereas 37% were actually genetically different (327). Four of 11 patients in the latter group had undergone keratoplasty between recurrences, but otherwise no other risk factor could be identified for infection with exogenous strains.
Infectious HSK in patients with AIDS is not entirely identical to that encountered in the immunocompetent patient. It has been reported that the incidence of HSV infection was no greater than in the general population, but recurrences of herpetic disease were more frequent in the HIV-positive population, with some patients having two to three recurrences over an average period of 17 months (183,299,328). These recurrences also tended to be lengthier than the initial episodes, with a clinical course requiring 2 to 4 weeks of topical therapy and a mean healing time of 3 weeks. There is little to no stromal infiltration under these ulcers, presumably as a result of the immunosuppressed
state of the patients. Immunocompetent patients usually had a recurrence-free interval of at least 18 months (295,308,314). Other reports on HIV-positive patients and patients with eczema noted that they benefitted from the use of oral acyclovir in more rapid resolution of their recurrent episodes of infectious disease (299,329).
state of the patients. Immunocompetent patients usually had a recurrence-free interval of at least 18 months (295,308,314). Other reports on HIV-positive patients and patients with eczema noted that they benefitted from the use of oral acyclovir in more rapid resolution of their recurrent episodes of infectious disease (299,329).
Hodge and Margolis have reported a large, controlled study on ocular HSV in HIV-positive patients (330). There were 1800 HIV-positive patient visits and 48,200 HIV-negative patient control visits. Although they also found no increased incidence of HSK, unlike earlier reports, there was no significant difference between HIV-positive and control patients in lesion type (epithelial and stromal), lesion location (peripheral vs. central), response time to topical trifluridine or oral acyclovir therapy (HIV-positive = 17 days; HIV-negative = 18 days), ultimate visual outcome, and time to first recurrence (347 days for HIV-positive vs. 321 days for HIV-negative). Only the recurrence rate was significantly higher in HIV-positive patients, being on average 1/587 days for HIV-positive patients and 1/1455 days for HIV-negative patients.
The pathogenesis of the branching ulceration typical of herpes epithelial keratitis has never been elucidated. The popular concept that it is related to neuronal distribution has been disproved (331). The pattern may simply be a function of viral linear spread by contiguous cell-to-cell movement. In HSV keratoplasties, the epithelium may occasionally heal in a superficial hypertrophic dendriform epitheliopathy pattern that may be confused with infectious dendritic ulcers. These lesions are sterile, however, and refractory to all treatment except therapeutic soft contact lenses (332).
Ulcerations occasionally occur within 2 mm of the limbus. These tend to be much more resistant to antiviral therapy than are central herpetic infections. Corneal sensation may remain normal in these marginal ulcers. These lesions must be differentiated from staphylococcal immune marginal ulcerations because the latter respond to steroid therapy and the former may worsen with such therapy.
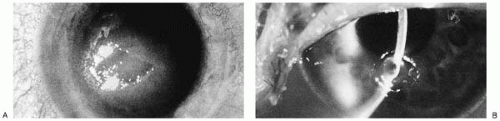 FIGURE 14-6. A: Thick-edged, sterile herpes simplex virus (HSV) trophic mechanical ulcer over active immune disciform keratitis. B: HSV trophic ulcer persisting over several months with subsequent melting (thinning) down to Descemet’s membrane (descemetocele) and impending perforation. |
Stromal reaction is usually absent or mild and confined to the anterior layers in milder epithelial infections. On occasion, however, even mild epithelial infection may be associated with notable stromal edema and iritis. These eyes are more likely to go on to chronic recurrent immune disease and scarring, resulting in visual loss. Concurrent or future stromal disease may be minimized, as well as the healing rate enhanced, by gentle debridement of the infected epithelium with a sterile cotton-tipped applicator before instituting antiviral chemotherapy (110,333). Such debridement is thought to remove much of the immune-inciting antigen that could penetrate to deep stromal layers.
Trophic (Metaherpetic) Ulceration. Occasionally, despite adequate antiviral therapy, epithelial ulcers do not completely heal or, having healed, break down again in an ovoid or dendritiform pattern. This is called trophic, or “metaherpetic,” keratopathy. This chronic sterile ulceration is secondary to interacting adverse factors, including impaired corneal innervation, abnormal tear film stability, and damaged basement membrane (125,334). Corneal epithelial cells normally lie on top of their basement membrane attached by hemidesmosomes, rather than interdigitating with the membrane. If the basement membrane is damaged during the infection or if the tear film lubricates poorly, the basement membrane takes at least 12 to 15 weeks to repair itself and thus slows adequate healing of the overlying epithelium (334). Gundersen’s classic study on the occurrence of trophic ulcers in several hundred patients with herpes revealed that trophic ulcers developed in 17% of patients receiving no therapy at all (335). Twenty-six percent treated with partial 10% iodine scrubs of the affected epithelium and 38% of those undergoing total epithelial removal with iodine scrubs acquired this condition.
Clinically, a trophic ulcer may be distinguished from an actively infected viral ulcer by the appearance of its edge. Trophic ulcers have gray, thickened borders formed by heaped-up epithelium unable to move across or adhere to the damaged ulcer base (Fig. 14-6A). In contrast, actively infected ulcers have discrete, flat edges that may change
their configuration as the ulcer erodes newly infected epithelium. Persistence of trophic ulceration over several weeks or months poses a threat to the integrity of the globe. The longer the ulcer is present, the greater the chance of collagenolytic activity with subsequent stromal melting (thinning) down to Descemet’s membrane, and perforation (Fig. 14-6B). This is particularly true in male patients and if the ulcer is located in the central cornea away from peripheral vascularization, which scars but heals (336,337).
their configuration as the ulcer erodes newly infected epithelium. Persistence of trophic ulceration over several weeks or months poses a threat to the integrity of the globe. The longer the ulcer is present, the greater the chance of collagenolytic activity with subsequent stromal melting (thinning) down to Descemet’s membrane, and perforation (Fig. 14-6B). This is particularly true in male patients and if the ulcer is located in the central cornea away from peripheral vascularization, which scars but heals (336,337).
Stromal Disease
Stromal disease may be divided into four to five categories according to currently accepted pathogenetic mechanisms: antigen-antibody-complement (AAC)-mediated immune disease characterized by viral interstitial keratitis, immune rings, and limbal vasculitis; and delayed hypersensitivity immune disease characterized by disciform edema/endotheliitis. The latter two are considered two separate categories by some authorities (i.e., disciform keratitis and endotheliitis). Endotheliitis is often associated with elevated intraocular pressure (23,297,338, 339, 340).
Interstitial Keratitis, Immune Rings, and Limbal Vasculitis. All three forms of herpetic AAC-mediated disease are thought to be due to immune complex hypersensitivity (125,184,341,342). The interstitial keratitis is a necrotizing keratitis (unlike disciform disease, which is nonnecrotizing). Interstitial keratitis presents clinically as single or multiple areas of dense, white infiltrates (Fig. 14-7). After several weeks of smoldering, deep leashes of vessels often move in as if in pursuit of the infiltrate(s). These lesions may remain small and focal or become so severe as to give the cornea a red (vessels) and white (interstitial keratitis) mottled pattern on unaided view. They resemble bacterial or fungal infiltrates, but are far more indolent and usually run a course of many months.
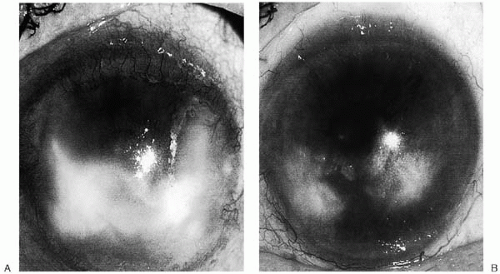 FIGURE 14-7. A: Dense, white, necrotic herpes simplex virus interstitial keratitis with 360 degrees of neovascularization. B: Same eye 1 year later after slowly tapered topical steroid therapy. |
A number of studies have reported presence of viral particles in HSK. We have reported two patients with ulcerative necrotizing HSK who despite months of topical and systemic antiviral therapy had numerous intact virions noted in the stromal keratocytes and extracellular lamellae on electron microscopic examination of the keratoplasty buttons (153). Using immunohistochemical stainings for HSV-1 antigens, Holbach et al. reported that 91% of 22 keratectomy specimens from patients with ulcerative necrotizing keratitis displayed HSV antigens (343). These antigens were located primarily in stromal keratocytes and the extracellular stroma, and less commonly in the endothelial and epithelial cells. Conversely, only 11% of 36 keratectomy specimens from patients with nonulcerative, nonnecrotizing keratitis or disciform stromal scarring had similar viral antigen staining. Histopathologic studies on a perforated graft with necrotizing stromal keratitis revealed that the endothelium and keratocytes contained viral inclusion bodies and antigens, indicating that a productive endothelial and stromal herpes infection can lead to graft failure and necrotizing stromal disease (344). Interstitial HSK is the most likely form of ocular herpes to recur in a new graft (153,345,346).
Indeed, molecular biologic studies on both animal models and human keratoplasty buttons have also provided evidence of a persistent or latent infection in peripheral
nonneural tissues, including the cornea (347, 348, 349). The discovery of live virus or viral antigen throughout all corneal layers in up to 25% of patients with herpetic keratitis substantiates the view that some stromal disease is due to active infection or a combination of infection and the immune processes discussed later (153,344,346,350).
nonneural tissues, including the cornea (347, 348, 349). The discovery of live virus or viral antigen throughout all corneal layers in up to 25% of patients with herpetic keratitis substantiates the view that some stromal disease is due to active infection or a combination of infection and the immune processes discussed later (153,344,346,350).
Immune (Wessely) rings in the anterior stroma and limbal vasculitis are thought to be two other clinical manifestations of AAC-mediated disease (351, 352, 353, 354, 355, 356). Limbal vasculitis is usually focal, although one or more quadrants may be involved in an edematous, hyperemic reaction sufficiently severe to cause dellen formation. There is little to no tendency for these vessels to invade the cornea in the absence of associated interstitial keratitis (Fig. 14-8A). The immune ring does not seem to incite major neovascularization but, not uncommonly, patches of interstitial keratitis may be associated with the ring and stimulate the growth of vessels (Fig. 14-8B and C).
Disciform Keratitis. Disciform edema of the cornea is a well-known adverse consequence of ocular herpes. It is categorized by some authorities as an entity separate from endotheliitis and by others as disciform keratitis/endotheliitis. It is currently believed that the pathogenesis of this lesion is a delayed hypersensitivity reaction to HSV-antigenic changes of the surface membranes of stromal cells and to the antigenically provocative residue of the viral invasion. The reaction itself is characterized histopathologically by a mixture of sensitized lymphocytes, plasma cells, macrophages, and neutrophils, as described later in the section on Immune Defense Mechanisms, under Pathogenesis (346,351,357).
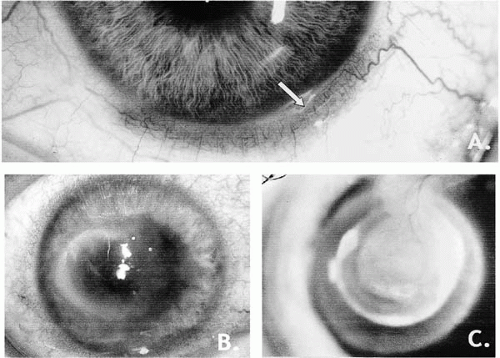 FIGURE 14-8. A: Herpes simplex virus (HSV) limbitis (arrow) with local edema and hyperemia due to local Arthus reaction. B, C: Single and double HSV immune rings in stroma. Double ring has a deep leash of vessels feeding to central cornea. |
Clinically, the milder to moderate forms of disciform disease present as focal, often disc-shaped stromal edema (Fig. 14-9). There is no necrosis or neovascularization, but there may be fine keratic precipitates attacking the endothelium just in the involved area, and there may be iritis. In some cases there is no anterior chamber reaction, yet keratic precipitates are present, which suggests that these programmed lymphocytes are particularly attracted to the focal corneal antigen in the endothelium or deep stroma. In
the more severe cases of disciform disease, the stromal edema is more diffuse, Descemet’s folds are present, and deep and superficial neovascularization may occur. Focal bullous keratopathy may develop, which indicates both that the endothelium is severely compromised by the inflammatory attack and that an abnormal amount of fluid is being driven into the cornea by hydrostatic pressure. In the most severe cases, the bullae rupture, and the cornea may become ulcerated with subsequent necrosis and melting. In moderate to severe cases of disciform disease, iritis is almost invariably present.
the more severe cases of disciform disease, the stromal edema is more diffuse, Descemet’s folds are present, and deep and superficial neovascularization may occur. Focal bullous keratopathy may develop, which indicates both that the endothelium is severely compromised by the inflammatory attack and that an abnormal amount of fluid is being driven into the cornea by hydrostatic pressure. In the most severe cases, the bullae rupture, and the cornea may become ulcerated with subsequent necrosis and melting. In moderate to severe cases of disciform disease, iritis is almost invariably present.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


