Tumors of Primitive Mesoderm, Striated Muscle, Adipose Tissue, and Smooth Muscle
Embryonal Rhabdomyosarcoma
This malignant tumor in the orbit was once considered a rare tumor of striated muscle. The present consensus holds that, in most patients, the usual source of the tumor is not adult striated muscle. Instead, the tumor recapitulates the primitive, multipotential nature of embryonal tissue. This characteristic is compatible with the high incidence of the neoplasm in children and adolescents and prompted the name embryonal rhabdomyosarcoma.
The tumor is not uncommonly found in neonates and, also, occasionally in adults. The revised histogenesis explains the changes in the incidence, treatment, and course of the lesion that have occurred during our 50-year tumor survey. References pertaining to the tumor have also increased manyfold in this interval.
Incidence
During the 50-year period 1948 to 1997, we collected information on 54 orbital rhabdomyosarcomas (26 primary, 27 secondary, one metastatic). When we omit the one metastatic tumor from the data, the remaining 53 rhabdomyosarcomas were 2.9% of our total 1,795 tumors (Table 3.3). There were 35 males and 18 females, nearly a 2:1 ratio. The age range of this sample was 1 to 39 years, with an average of 12.3 years and a median of 9 years. These age data confirm the generally held view that rhabdomyosarcoma is the most common orbital malignancy in the first two decades of life. Histologically, 48 tumors were embryonal, 2 alveolar, and 2 pleomorphic. One was not subclassified, and the paraffin block was no longer available for a second review.
Oberlin et al. (2001) published the experience of four international collaborative groups—Rhabdomyosarcoma Study Group, International Society of Paediatric Oncology Sarcoma Committee, German Collaborative Soft Tissue Sarcoma Group, and Italian Cooperative Soft Tissue Sarcoma Group. Collectively, these groups studied 306 patients with orbital rhabdomyosarcoma. The median age of these patients was 6.8 years.
Clinical Features
Ninety-one percent (48 out of 53) of the orbital rhabdomyosarcomas in our 50-year survey were the embryonal type. Herein, we chiefly discuss the clinical features of this age-group.
The most common clinical features are a painless proptosis of one eye of rather sudden onset, soon followed by swelling of one or both eyelids, chemosis of conjunctiva, mechanical impairment of ocular motility in one direction of gaze, and nonaxial displacement of the eye.
As a rule, symptoms develop rapidly in young patients, with the affected eye and adnexa showing obvious progression by the day or week, rather than by the month. At onset, the puffy edematous eyelids show a bluish tinge that soon changes to a reddish purple hue giving way to an alarming red color that suggests an inflammatory process (see Fig. 7.1).
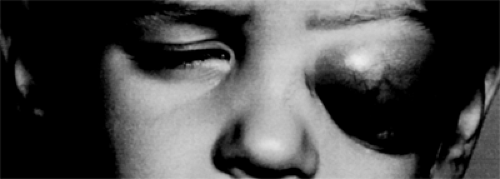 Figure 7.1 Red, slightly violaceous, edematous eyelids concealed a proptosed left eye of 3 weeks’ duration in a 4-year-old boy. The marked edema of the eyelids was disproportionate to the short duration of the process. (See Color image.) |
There is no increase in local heat, however, and the process is painless unless a number of weeks have elapsed since onset. Affected patients are not febrile nor do the children look sick, as would be the case in children with orbital cellulitis, neuroblastoma, or Langerhans histiocytosis, which occur in this age-group. Often, the concerned parents of an affected child attribute the puzzling proptosis and swollen eyelid to trauma, although such trauma is either not well substantiated or is so trivial that it is not etiologically important. The swollen eyelid may partially conceal the proptosis and resist palpation. Grossly, the tumors are well defined.
The embryonal rhabdomyosarcoma is noted for unusual presentations as well as association with other benign and malignant neoplasms and other disorders of children. Onderoglu et al. (1999) described multiple masses of a solid tumor in a fetus at the 33rd week of gestation, involving the liver, orbit, and maxillary sinus and detected by ultrasonography. The presumptive diagnosis was rhabdomyosarcoma, which was later confirmed. Chams et al. (1988) described a case in a 1-day-old infant.
Hadjistilianou et al. (2002) reported two infants, aged 2 months and 11 months, with orbital rhabdomyosarcoma associated with neurofibromatosis type 1. Muwakkit et al. (2002) noted a simultaneous occurrence of Wilms tumor and rhabdomyosarcoma in two patients, and Armstrong and Sclar (1999) described a 16-year-old girl with orbital rhabdomyosarcoma who developed bloody diarrhea during chemotherapy. She was found to have a familial adenomatous colonic polyposis.
Metastatic rhabdomyosarcoma may also involve the orbit. Amato et al. (2002) reported metastasis to the right orbit in a 29-year-old man 18 months after the initial tumor in the left orbit had been treated with chemotherapy and radiotherapy. Simpson et al. (1999) noted a metastatic tumor presenting as an isolated lateral rectus muscle restriction. The one case in our tumor survey metastasized from the testes of a 29-year-old. Fekrat et al. (1993) described a 27-year-old woman with metastasis of an alveolar rhabdomyosarcoma to the orbit.
Earlier we noted the tendency of embryonal rhabdomyosarcoma to occur in individuals older than the second decade. Our 50-year survey included three patients in the fourth decade with rhabdomyosarcoma. One patient was 36 years old, and two others were each 39 years old. Other individuals in this age range have been reported in the literature.
The third through seventh decades are the age range for pleomorphic “striated muscle” rhabdomyosarcoma of adults. We have only one patient with the pleomorphic tumor in our survey, and our experience with the multifaceted clinical features of this histologic type is too limited to write a comprehensive review. Readers who encounter a pleomorphic rhabdomyosarcoma may be interested in the review of Furlong et al. (2001). These authors published a thorough clinicopathologic study of 38 cases from multiple anatomic areas, including one orbital case. There were 28 men and 10 women with a mean age of 52 years and a median of 54 years. They conclude that the tumor is a high-grade sarcoma with an aggressive clinical course. The spindle cell variant also tends to affect adults and has a good prognosis.
Imaging Aspects
Computed tomography (CT) scan of this tumor is important because it shows the tumor’s size, configuration, location, and the presence or absence of bone destruction. These tumors may occur anywhere in the orbital space but favor the superonasal quadrant. Small tumors are usually fairly well defined, may even appear circumscribed, and show mild to moderate contrast enhancement (see Fig. 7.2).
Large tumors have irregular contours and show marked contrast enhancement. The degree of enhancement is probably related to the volume of blood supply.
Large tumors have irregular contours and show marked contrast enhancement. The degree of enhancement is probably related to the volume of blood supply.
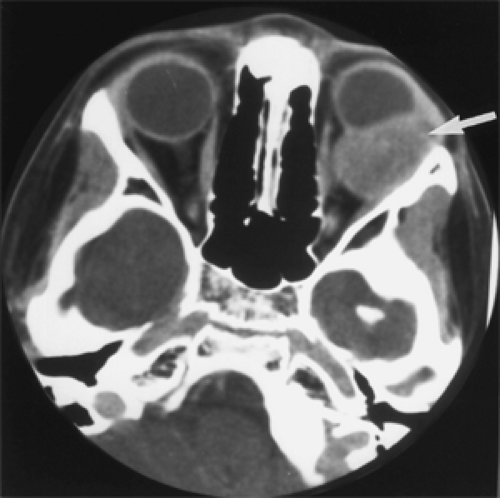 Figure 7.2 Axial computed tomography scan of a primary rhabdomyosarcoma shows a homogeneous, slightly enhancing mass (arrow) in the left posterior, superolateral orbit of a 10-year-old boy who was symptomatic for only 2 weeks. The tumor measured 30 × 25 × 20 mm. |
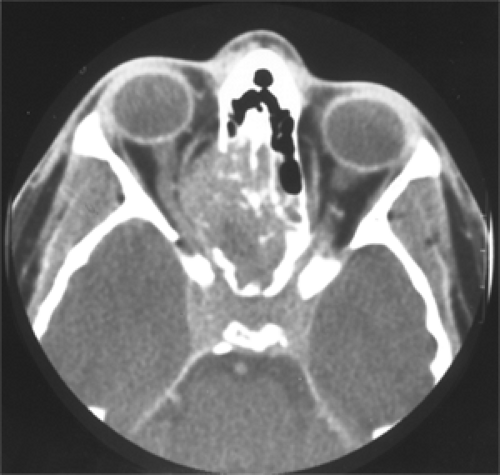 Figure 7.3 Axial computed tomography scan shows a large soft tissue mass in the right ethmoid sinus with destruction of the adjacent medial orbital wall, extension into the orbital cavity, and spread to the opposite ethmoid of 2 months’ duration in a 17-year-old boy. |
If bone destruction is present, the medial orbital wall and thin portion of the lateral wall are most often affected. Osseous involvement is always present when secondary tumors from the nasal passage, a paranasal sinus, or the intracranial vault invade the orbit (see Fig. 7.3). Bone destruction frequently accompanies recurrence of a primary orbital rhabdomyosarcoma and was present at the time of presentation in about one third of our patients with primary orbital tumors. A primary rhabdomyosarcoma with osseous or paranasal involvement is staged in a more serious category for the purpose of the Intergroup Rhabdomyosarcoma Study compared with a tumor without bone encroachment.
Magnetic resonance imaging (MRI) shows an isotense or slightly hypotense lesion compared with brain on a T1-weighted scan. T2-weighted scan shows a hypertense signal. Radiologists Mafee et al. (1998) believe a CT scan or MRI “baseline” scan is essential after completion of therapy. The scan is specific for showing recurrence of tumor that otherwise may not be clinically suspected.
Pathology
Grossly, the tumor is soft and varies in color from grayish white to yellow tan or reddish tan. It may be obviously infiltrative (see Fig. 7.4) or deceptively circumscribed (see Fig. 7.5). Although the latter tumor mass can often be removed intact despite its fragile consistency, the surrounding orbital tissue shows microscopic spread of tumor beyond the line of circumscription.
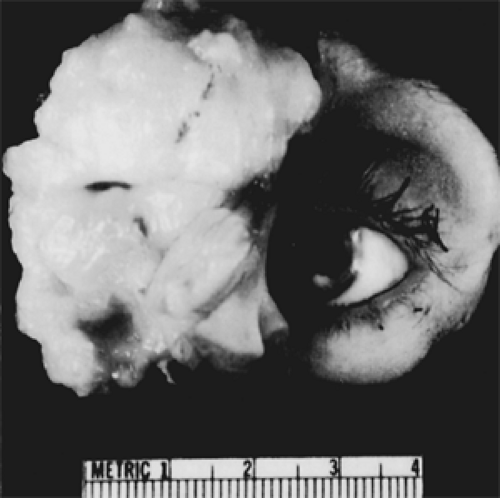 Figure 7.4 Exenteration specimen of an embryonic rhabdomyosarcoma shows a large, fleshy, grayish white, infiltrative tumor. (See Color image.) |
Various cell types and growth patterns characterize this neoplasm. The cell morphology may range from small, undifferentiated round cells to increasingly differentiated
types, including spindle-shaped cells, strap cells, racquet cells, tadpole cells, spider cells, and giant cells. In general, cross-striations are seen only in better-differentiated tumors. The histologic types are subclassified as embryonal, alveolar, and pleomorphic. The term botryoid is applied to the gross grape-like appearance of the rare submucosal mass that may appear in the epibulbar tissues. This is usually of the embryonal type.
types, including spindle-shaped cells, strap cells, racquet cells, tadpole cells, spider cells, and giant cells. In general, cross-striations are seen only in better-differentiated tumors. The histologic types are subclassified as embryonal, alveolar, and pleomorphic. The term botryoid is applied to the gross grape-like appearance of the rare submucosal mass that may appear in the epibulbar tissues. This is usually of the embryonal type.
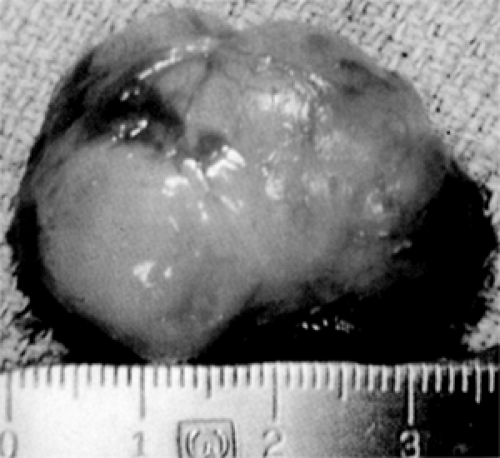 Figure 7.5 This 35-mm embryonal rhabdomyosarcoma is reddish yellow, slightly lobulated, soft, and moderately circumscribed, apparently removed intact from a 4-year-old boy. However, residual tumor was visible microscopically in surrounding orbital tissue. (See Color image.) |
The embryonal type (see Fig. 7.6A) shows some combination of undifferentiated small round cells admixed with cells attempting rhabdomyoblastic differentiation. These latter cells may be small and round with deeply eosinophilic cytoplasm that is acidophilic or spindle-shaped with hyperchromatic nuclei and acidophilic cytoplasm. Other cells include elongated strap cells with one or two nuclei arranged in tandem. Intermingling streams of spindle cells may be associated with a myxoid stroma. Intracellular glycogen, which can be demonstrated in most rhabdomyosarcomas, is sparse in this poorly differentiated embryonal form. The spider cells are examples in which the intracellular glycogen has been moved by fixation.
Those embryonal tumors in which fascicles of spindle cells predominate have a higher degree of differentiation. Within the elongated strap cells, one or two nuclei are arranged in tandem, and the cytoplasm is acidophilic. Well-marked cross-striations may be visible with hematoxylin and eosin stain.
The alveolar type, which is the second most common type, is so called because the framework is similar to that of the lung. The tumor cells are similar to those found in the embryonal form and are present within the alveolar spaces and cling to the fibrous connective tissue septa (Fig. 7.6B). It is not uncommon to find an alveolar pattern as a small focus in a tumor that is predominantly of the embryonal type. The alveolar subtype is thought to have the worst prognosis, regardless of its location or stage.
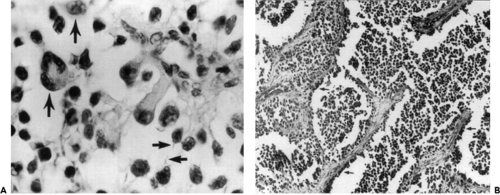 Figure 7.6 Embryonal rhabdomyosarcoma shows a variety of cell types. A: Note a large strap cell (center) with a large nucleus at the lower pole, a spiderweb cell (upper left arrow), and a large, multinucleated cell (left center arrow). Many of the cells had a wispy extension of cytoplasm (lower right horizontal arrows), suggesting a tadpole-like contour. The size and shape of the hyperchromatic nuclei differ (original magnification × 600). B: Another area of the same tumor shows alveolar features. Note the orderly row of cells clinging to the connective tissue septa along the periphery of the cell clusters (arrows) and the loose arrangement of the cells in the center of the cell clusters (original magnification × 90). |
Management
As chemotherapy has improved, attempts have been made to spare radiation therapy in a select group of patients (Alvarez Silvan et al., 1996; Lackner and Urban, 1997). Nowadays, however a patient living in the United States who presents with an orbital tumor that, on biopsy, is an embryonal rhabdomyosarcoma may become part of a therapeutic protocol of some state, regional, or national rhabdomyoma study group. The orbital lesion has a more favorable prognosis compared with rhabdomyosarcoma in many other organs and anatomic sites because of the absence of multiple, systemic foci of tumor.
The Mayo Clinic Section of Pediatric Oncology recommends a staging workup, which includes “CT or MRI at the primary site, evaluation of lymph nodes, chest CT scan, bone scan, and bilateral marrow aspirant biopsy.” If there is no metastatic spread of the tumor, the patient is staged into one of three clinical groups: Group 1, complete surgical excision of tumor with no microscopic residual tumor; group 2, microscopic residual tumor; and group 3, gross residual disease. If the tumor has undergone an incisional biopsy and then been debulked, it belongs in group 2. If the tumor is large, frankly infiltrative, and not debulked after biopsy, it belongs in group 3.
In the past we encountered cases that were operated on within 3 weeks of onset; the tumor was small, located in the retrobulbar space, and grossly circumscribed. With careful dissection and the use of cryoextraction, the surgeon may believe the tumor has been removed intact, but the tumor’s circumscription may be deceiving. Bits of tissue from the adjoining retrobulbar tissue may show microscopic spread on further microscopic study. True “complete excision” of an orbital rhabdomyosarcoma is probably rare, except for an exenteration.
No radiotherapy is used for group 1 cases. This group implies clear surgical margins. Radiotherapy of 36 Gy is administered for group 2. For group 3, 45 Gy of radiotherapy is given at week 3. Two drugs, vincristine and actinomycin, are administered initially. Cytoxan may be added to the regimen if lymph nodes are involved. A more aggressive treatment protocol is needed for an alveolar rhabdomyosarcoma of the orbit.
Rhabdoid Tumor
Rhabdoid (from the Greek, phabdo-eides, like a rod, rod-shaped) is usually a primary tumor of the kidney in children <1 year of age. Basically, it is another member of the clan of poorly differentiated, mesodermal, small, round cell tumors. Initially, Beckwith and Palmer (1978) thought it was a sarcomatous variant of Wilms tumor and named it rhabdomyosarcomatoid tumor because of its light microscopic resemblance to embryonal rhabdomyosarcoma. Later (Haas et al., 1981), on the basis of ultrastructural study of 11 examples available from the National Wilms Tumor Study, the neoplasm was considered a distinct entity separate from rhabdomyosarcoma and given the name rhabdoid tumor.
Subsequently, tumors with a histologic appearance similar to that of rhabdoid tumors arising in the kidney have been described in virtually every extrarenal anatomic site. Carcinomas of various types may also have rhabdoid features. The term extrarenal rhabdoid tumor as it pertains to soft tissue should be used for tumors with predominant rhabdoid morphology and in which no other clear line of differentiation can be documented (Weiss and Goldblum, 2001).
The presentation of a rhabdoid orbital tumor is so similar to a rhabdomyosarcoma (see Fig. 7.7) that it is impossible to differentiate them at this early stage. A primary rhabdoid tumor of the orbit is very rare. Rootman et al. (1989) seem to be the first to report such a case. This was a 6-week-old boy with rapidly progressive proptosis since the age of 1½ weeks. The proptosis of the child’s left eye measured 10 mm. CT scan showed a large, nonenhancing retrobulbar mass with expansion of the orbital walls and extension of tumor into the superior and inferior orbital fissures. The tumor was debulked with a suction fragmentation device. The patient was treated with a combined course of radiotherapy (41 Gy) and chemotherapy. The child was alive and well without recurrence or second tumor 24 months later. The second case was a 47-year-old man (Johnson et al., 1991) who presented with increasing hyperopia and monocular diplopia of 3 months’ duration. A circumscribed, firm tumor within the sheath of the left lateral rectus muscle was resected. Postoperatively, he received 50 Gy of radiotherapy in 25 fractions. There was no recurrence of tumor 18 months postoperatively.
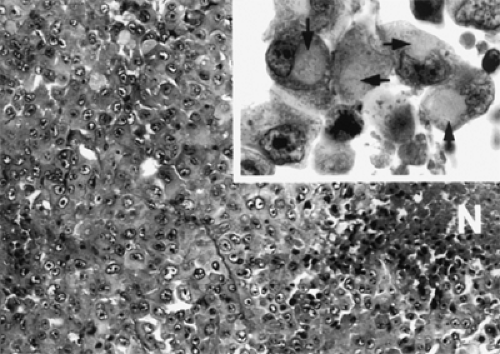 Figure 7.7 This recurrent rhabdoid orbital tumor is composed of poorly differentiated cells with copious cytoplasm continuing filamentous inclusions with prominent nucleoli. N marks the focus of necrosis (hematoxylin and eosin, original magnification × 50). Inset: Arrows denote filamentous cytoplasmic inclusions (hematoxylin and eosin, original magnification × 250). (From Gunduz K, Shields JA, Eagle RC Jr, et al. Malignant rhabdoid tumor of the orbit. Arch Ophthalmol. 1998;116:243–246 , with permission.) |
A third patient, a 3-year-old girl (Gunduz et al., 1998), did not fare as well as the two preceding patients. The biopsy diagnosis was made after a 3-week history of proptosis of the right eye. Despite initial chemotherapy and radiotherapy, she had massive orbital recurrence 6 months later, and an exenteration was performed. Aggressive chemotherapy with a combination of vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide phosphate was continued. At 17 months’ follow-up, orbital debulking surgery with externalization of the maxillary sinus was performed because of massive tumor recurrence. The child died 28 months after her initial diagnosis from tumor invasion of the central nervous system.
A fourth case (Niffenegger et al., 1992) was a 50-year-old man with a rapidly growing mass in the area of the right lacrimal gland. A wide local excision of a yellow mass was performed. A workup for metastases was negative, and orbital exenteration was then performed. Five weeks after exenteration, 45 Gy of radiotherapy was delivered in 25 fractions over 40 days. Six weeks after completion of radiotherapy, the patient was started on systemic chemotherapy, including cycles of actinomycin, doxorubicin, cyclophosphamide, and vincristine. Fifteen
months later, the patient had no evidence of either local recurrence or distant metastases.
months later, the patient had no evidence of either local recurrence or distant metastases.
Stidham et al. (1999) reported a primary malignant rhabdoid tumor of the orbit in a neonate. Walford et al. (1992) described an intraorbital rhabdoid tumor arising in the right orbit of a 3-year-old boy previously treated for bilateral retinoblastoma. The patient first presented at the age of 2 months. The left eye was enucleated, and the right orbit was treated with radiotherapy (lateral field, 45 Gy). Recurrent tumor was noted 27 months later in the right orbit. The patient was treated with chemotherapy and additional radiotherapy but failed to respond. He died as the result of intracranial tumor progression.
Imaging Aspects
With CT scan, the cases of Rootman et al. (1989) and Gunduz et al. (1998) showed a poorly defined, homogeneous, nonenhancing mass in the retrobulbar space. If the tumor is quite large, there may be thinning of the orbital bones with some expansion of the orbit.
Pathology
These tumors are highly malignant with rapid growth, and the mortality rate is high. Grossly, the orbital tumor appears pale tan to white and is likely to be circumscribed to some degree. However, if it has been present for >3 to 4 weeks, it may be widely infiltrative.
The light microscopic features are sheets or nests of round, oval (epithelioid-like) cells with eosinophilic cytoplasmic filaments, eccentric nuclei, and “owl eye” (Spencer, 1996). The hyaline-like globules of cytoplasmic, intermediate filaments may be so large as to indent the nuclei. Vimentin staining is usually positive. Other immunostaining may be variable.
Management
Once the incisional biopsy is positive for rhabdoid tumor, an oncologist should be contacted quickly to oversee the patient’s staging workup. When the workup is completed, the oncologist may recommend one of the current protocols of therapy used by the Intergroup Rhabdomyosarcoma Study. The therapy involves some combination of chemotherapy, surgical excision, and radiotherapy. However, too few cases of orbital rhabdoid have been treated and followed up over an adequate period of time to have a standard, uniform consensus as to sequence of treatment entities, dosage, and duration of therapy.
Rhabdomyoma
Terminology
Rhabdomyoma is a benign neoplasm of striated muscle that occurs most commonly in the heart of infants. Extracardiac rhabdomyomas are divided into four different types on the basis of histologic differences, the patient’s age, and the organ or area of the body affected. Ophthalmologists need to be concerned only with the adult and fetal types.
Neoplasms of striated muscle relevant to this discussion include, in ascending order of their histologic differentiation toward normal adult striated muscle, embryonal rhabdomyosarcoma, fetal rhabdomyoma, pleomorphic rhabdomyosarcoma, and adult rhabdomyoma, the best differentiated (Knowles and Jakobiec, 1975). Some authors regard fetal rhabdomyoma as the benign counterpart of embryonal rhabdomyosarcoma and adult rhabdomyoma as the benign counterpart of pleomorphic rhabdomyosarcoma. The qualifier “adult” seems to be based on this classification. However, in this regard, it is a misnomer because this well-differentiated tumor also occurs in children.
Clinical Features
The so-called adult rhabdomyoma presents as a slow-growing, painless, solitary, round or polypoid mass in the head and neck region. Here it principally affects the pharynx or larynx, causing hoarseness or difficulty in swallowing (Weiss and Goldblum, 2001). Two cases of orbital involvement have been well-authenticated (Knowles and Jakobiec, 1975; Hatsukawa et al., 1997).
The case described by Knowles and Jakobiec (1975) was an 8-year-old boy who presented with a 3-month history of a painless, firm, enlarging mass in the left upper eyelid. At surgery, an encapsulated, multilobulated mass was found arising from the medial rectus muscle. (The tumor’s attachment to the orbital muscle differentiates it from embryonal [undifferentiated] rhabdomyosarcoma in the same age-group.) Four months later, the patient returned with a pea-sized lesion overlying the insertion of the medial rectus muscle. This lesion was thought (mistakenly) to be an embryonal rhabdomyosarcoma, and an orbital exenteration was performed. Twenty-five years later, the patient was alive and free of recurrence.
The case described by Hatsukawa et al. (1997) was a 16-month-old boy with painless proptosis (3 mm) of the right eye of 1 month’s duration. On lateral orbitotomy, an engorged lateral rectus muscle was noted. The orbital mass was partially excised from its attachment to the muscle. One year after surgery, CT scan did not show any regrowth of tumor.
Kapadia et al. (1993) studied 24 fetal rhabdomyosarcomas that presented as well-defined solitary masses arising within the soft tissue or mucosa of the head and neck and displayed a greater degree of maturation and a wider morphologic spectrum than previously recognized. In this summary of cases, one patient, an 18-year-old man, was listed as having an orbital mass associated with proptosis and decreased vision. No other clinical features of this case were stated. However, the histologic specimen, illustrated in the article, was removed from the periorbital area. This
case may be a primary tumor of the eyelid with secondary invasion of the orbital space. Kapadia et al. (1993) made an important observation; that is, fetal rhabdomyomas occur in a broader age range and histologic spectrum than previously recognized. Eleven tumors (46%) occurred in patients older than 15 years.
case may be a primary tumor of the eyelid with secondary invasion of the orbital space. Kapadia et al. (1993) made an important observation; that is, fetal rhabdomyomas occur in a broader age range and histologic spectrum than previously recognized. Eleven tumors (46%) occurred in patients older than 15 years.
Our 50-year tumor file includes no orbital rhabdomyomas.
Imaging Aspects
We have found only one CT scan of an orbital rhabdomyoma in the literature (see Fig. 7.8). This showed an irregularly contoured, somewhat mottled mass in the right orbital apex that extended forward along the medial and lateral rectus muscles. The mass did not enhance after contrast injection. A plain skull film did not show bony erosion (Hatsukawa et al., 1997).
Pathology
The so-called adult-type rhabdomyoma is a gray-yellow, fibrous mass composed of mature but disorganized striated muscle fibers with a mixture of collagen fibers. Cellular details are illustrated in Figure 7.9. There is no nuclear atypia. Metastases are absent. Immunohistochemically, the cells stain positively for myoglobin, desmin, and muscle-specific actin but are positive less commonly for vimentin and S-100 protein (Weiss and Goldblum, 2001).
According to Kapadia et al. (1993), so-called fetal rhabdomyomas of soft tissues are grossly circumscribed, soft, and gray white to tan pink with a mucoid glistening cut surface. The tumors are unencapsulated. Their histologic picture is quite different from adult-type rhabdomyomas. Overall, the fetal type shows a spectrum of skeletal muscle differentiation ranging from the “classic” immature tumors to those lesions with more advanced rhabdomyoblastic maturation. They refer to the latter group as intermediate differentiation, a degree that never completely recapitulates the appearance of normal adult skeletal muscle. The intermediate type is illustrated in Figure 7.10. This specimen is from the 18-year-old man described previously.
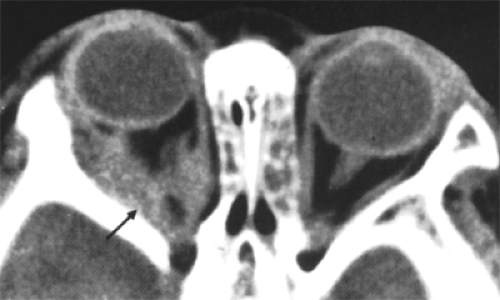 Figure 7.8 Axial computed tomography scan shows an irregular right retrobulbar mass (arrow). The tumor did not enhance after contrast injection. (From Hatsukawa Y, Furukawa A, Kawamura H, et al. Rhabdomyoma of the orbit in a child. Am J Ophthalmol. 1997;123:142–144 , with permission.) |
 Figure 7.9 Rhabdomyoma is composed of large, round polygonal cells with acidophilic, granular cytoplasm and large, vesicular, peripherally placed nuclei (trichrome, original magnification × 375). (From Knowles DM II, Jakobiec FA. Rhabdomyoma of the orbit. Am J Ophthalmol. 1975;80:1011–1018 , with permission.) |
The immature classic type is composed of an admixture of haphazardly arranged, slender skeletal muscle fibers with rare cytoplasmic cross-striations and undifferentiated round, spindled, serpentine cells in a myxoid stroma. All 24 specimens the authors examined had cross-striations.
Immunohistochemical stains were positive for myoglobin, desmin, and muscle-specific actin.
Immunohistochemical stains were positive for myoglobin, desmin, and muscle-specific actin.
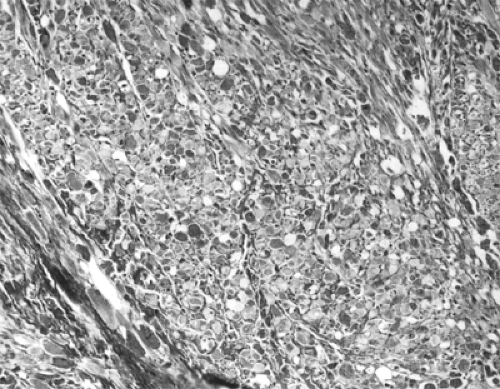 Figure 7.10 An “intermediate” fetal rhabdomyoma from the periorbital region shows a fascicular arrangement of cells with abundant eosinophilic cytoplasm and rare cytoplasmic vacuolation (original magnification × 75). (From Kapadia SB, Meis JM Frisman DM, et al. Fetal rhabdomyoma of the head and neck: A clinicopathologic and immunophenotypic study of 24 cases. Hum Pathol. 1993;24:754–765 , with permission.) |
Management
A guarded debulking or partial excision of the tumor seems to suffice, taking care to preserve, as much as possible, the affected extraocular muscle.
Malignant Mesenchymoma
Terminology
The present concept of mesenchymoma evolved from the interest of Stout (1948) in the morphology and classification of soft tissue sarcomas, particularly in children. Working only with the media of light microscopy and tissue culture (electron microscopy was in its infancy), he unified a number of poorly differentiated, unclassified sarcomas of mixed morphology and applied the simple term mesenchymoma to this new conglomerate of tumors. This name brought order into a disordered field of complex, compound names that had previously been used to designate the multiple histologic features of these sarcomas. It is likely that the term mesenchymoma was borrowed from an earlier publication of Klein (1932).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


