Tumors of Peripheral Nerve Sheath Origin
The origin of many tumors in this text is linked, wholly or in part, to mesoderm. However, nerve sheath tumors arise from neuroectodermal tissue. Ophthalmologists are principally interested in the benign proliferation of elements of the sheaths of branches of the cranial, motor, and sensory nerves that traverse the orbital space. These disorders are the solitary neurofibroma, neurofibromatosis type 1, and schwannoma. They have one feature in common, that is, a tendency to undergo malignant transformation. The malignant forms are lumped together under the broad term malignant nerve sheath tumor.
These four types of orbital nerve sheath neoplasms are the most protean and ubiquitous of the numerous tumor families in this text. The incidence of nerve sheath tumors among the total tumors in our 50-year survey (see Chapter 3) is 3.5% (63/1,795). In their various guises, these tumors may be associated with optic nerve glioma, buphthalmos, iris nodules, hamartomas of the choroid, posterior capsular cataract, asymmetry of the facial bones, hypoplasia of adjacent paranasal sinuses, dysplasia or dysgenesis of the sphenoid bone, and eyelid tumors.
Solitary Neurofibroma
Solitary neurofibroma may also be called isolated neurofibroma.
Incidence
Nine neoplasms in our series were the solitary type. The age at onset was known in eight of the patients, ranging from 20 to 58 years (median, 45.5 years). Sex incidence was equal. Two of these patients had more than one solitary lesion in the unilateral orbit (see Fig. 8.1). Multiple forms of this lesion have also been reported in the literature (Gurland et al., 1976; Krohel et al., 1985; Shields et al., 1990; Meyer and Wobig, 1992). Shields et al. (1990) encountered three separate neurofibromas in one orbit. The case described by Meyer and Wobig (1992) was unusual, a solitary neurofibroma in each orbit. This patient also had several features suggestive of multiple endocrine neoplasia type 2B. Multiple neurofibromas are usually associated with neurofibromatosis type 1 (NF1).
Clinical Features
The usual orbital location of a solitary neurofibroma is the supraorbital frontal branch of the trigeminal nerve. Second in frequency is the maxillary branch. Multiple tumors in patients also involve other nerves transversing the orbit (see Fig. 8.2). The tumor is slow-growing. The usual pattern of presentation is a progressive unilateral proptosis of 1 or more years’ duration. The tumor exerts a mass effect on the eye, and the eye’s displacement is opposite the growing mass. Initially there is no pain, but pain of variable degree develops as the tumor expands the covering perineurium and epineurium of the nerve. Visual acuity is little affected.
One of our patients, a 52-year-old woman, had quite a different presentation. A year before admission she had noted slight proptosis of the left eye. One month later, she had a sudden, marked reduction in vision in the affected eye. She was treated for optic neuritis, without effect. On presentation, vision was reduced to counting fingers associated with pallor of the left optic disk and a cecocentral scotoma. Her neurofibromas proved to be localized to the apex of the left orbit.
Imaging Aspects
The ophthalmologist may have some trouble differentiating between a solitary neurofibroma and a cavernous hemangioma on computed tomography (CT) scan. Both lesions have about the same degree of enhancement, are well outlined, have similar homogeneity, and are located in the retrobulbar space. However, on anteroposterior and
sagittal views, the neurofibroma is more rounded in shape and positioned more forward in the retrobulbar space. Indeed, the front edge of the neurofibroma may overlap the rear edge of the eyeball. Other features of a neurofibroma are noted in Figure 8.3.
sagittal views, the neurofibroma is more rounded in shape and positioned more forward in the retrobulbar space. Indeed, the front edge of the neurofibroma may overlap the rear edge of the eyeball. Other features of a neurofibroma are noted in Figure 8.3.
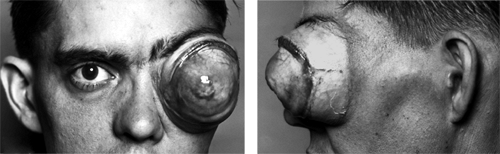 Figure 8.1 Front and side views of a huge neurofibroma in a 32-year-old man that developed slowly over a period of 15 years. The left eye was blind. The patient asked to have the orbital mass removed because of increasing pain. An exenteration was performed. The tumor measured 8 × 7 × 6 cm. |
Pathology
A gross specimen is illustrated in Figure 8.4. The basic unit of the localized neurofibroma is a myelinated or nonmyelinated peripheral nerve axon covered by intertwining Schwann cells, which are intimately associated with collagen strands and surrounded by a matrix of mucopolysaccharide, capillaries, and endoneural fibroblasts, all in varying ratios (see Fig. 8.5). This tissue complex causes a fusiform or oval intraneural expansion of the involved nerve. The tumor is circumscribed, is grayish white, and has a firm fibrous consistency. With low-power light microscopy, the distinctive feature is wavy interlacing bundles of collagen interspersed with spindle-shaped cells and separated by a mucopolysaccharide-rich material (see Fig. 8.6A). Under higher power, the mixture of spindle cells can be differentiated into endoneural fibroblasts and those with comma-shaped nuclei (Schwann cells). This arrangement may be compact and cellular or loose-textured
and myxoid in appearance. Capillarity is not a prominent feature of this tumor compared with the plexiform neurofibroma. The axonal component of these tumors can be identified with the Bodian stain (colloidal silver), and the matrix can be stained with Alcian blue (a copper-containing dye). A neurofibroma containing spherical tactile-like bodies is illustrated in Figure 8.6B. Immunohistochemically, the tissue stains positive for S-100 protein.
and myxoid in appearance. Capillarity is not a prominent feature of this tumor compared with the plexiform neurofibroma. The axonal component of these tumors can be identified with the Bodian stain (colloidal silver), and the matrix can be stained with Alcian blue (a copper-containing dye). A neurofibroma containing spherical tactile-like bodies is illustrated in Figure 8.6B. Immunohistochemically, the tissue stains positive for S-100 protein.
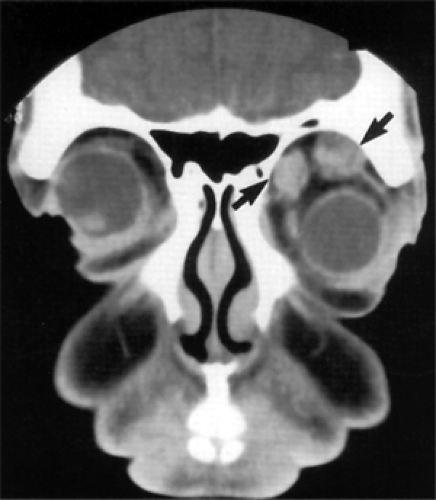 Figure 8.2 Multiple neurofibromas. Coronal computed tomography scan shows two circumscribed tumors (arrows) in the left orbit of a 42-year-old man. At orbitotomy, the masses were connected by a small isthmus of tissue. |
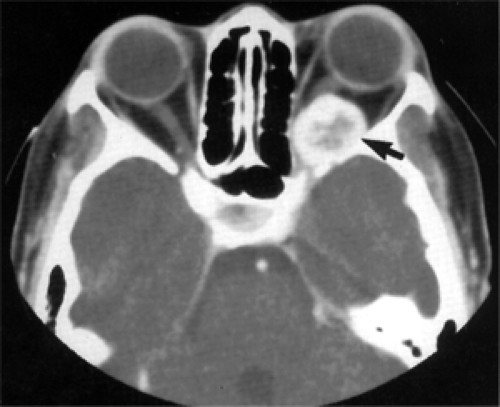 Figure 8.3 Axial computed tomography scan shows an enhancing circumscribed mass in the apex of the left orbit (arrow) with a hypodense center that remodeled and expanded orbital bone of a 52-year-old woman with proptosis of 1 year’s duration. Vision was reduced to counting fingers secondary to optic nerve compression. |
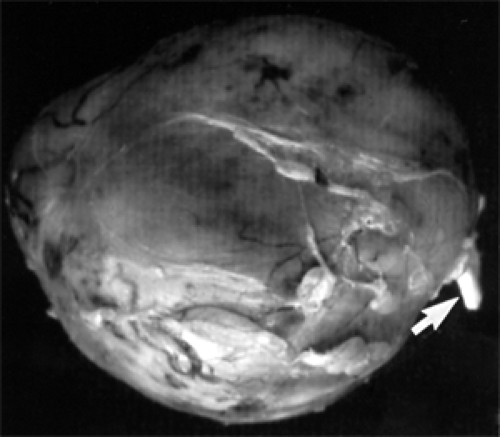 Figure 8.4 Gross specimen of a circumscribed, oval expansion of an orbital nerve. At the right (arrow) is the pigtail segment of normal nerve from which it arose. Specimen measured 2 × 2.5 × 2 cm. |
Here, we should mention the diffuse neurofibroma that many pathologists consider a distinct type of nerve sheath tumor. Histopathologically, it has cellular components similar to those of the solitary neurofibroma, but it is an infiltrating, extraneural neoplasm that expands along connective tissue septa and intercellular spaces, replacing orbital fat and permeating extraocular muscle (see Fig. 8.7). Its clinical behavior and management are more akin to those of a schwannoma. From a practical standpoint, other authors choose to combine these tumors in their discussion of schwannoma.
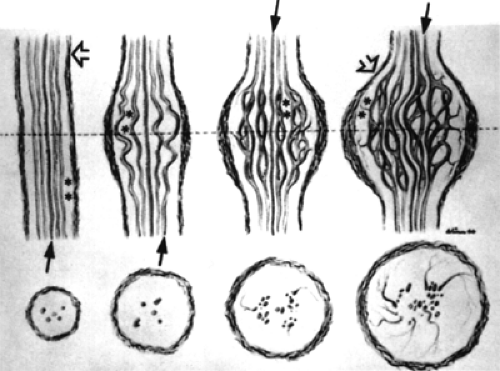 Figure 8.5 Diagrammatic representation of progressive phases (left to right) of the tumor’s growth that spreads the Schwann cell cylinders (arrows) apart. Perineurium (arrowheads) is thickened. Asterisks indicate endoneural tissue. Cross-sections through the level of the dotted line are shown at the bottom of the illustration. (From Harkin JC, Reed RJ. Tumors of the peripheral nervous system. In: Atlas of tumor pathology, Second Series, Fascicle 3, Washington, DC: Armed Forces Institute of Pathology; 1969 , with permission.) |
Management
The solid makeup of the solitary tumor facilitates its intact removal. If it can be removed intact, it does not recur.
Excision is reasonably free of bleeding. In the surgical management of nerve sheath tumors, care should be taken to identify the involved nerve as a sensory nerve rather than a motor nerve before resecting it.
Excision is reasonably free of bleeding. In the surgical management of nerve sheath tumors, care should be taken to identify the involved nerve as a sensory nerve rather than a motor nerve before resecting it.
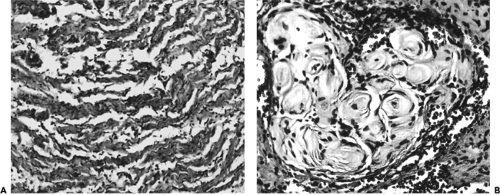 Figure 8.6 A: Wavy bundles of collagen interspersed with Schwann cells and endoneural fibroblasts and separated by a mucinous-myxoid matrix (original magnification × 130). B: Some areas may contain spherical tactile-like corpuscles (original magnification × 200). |
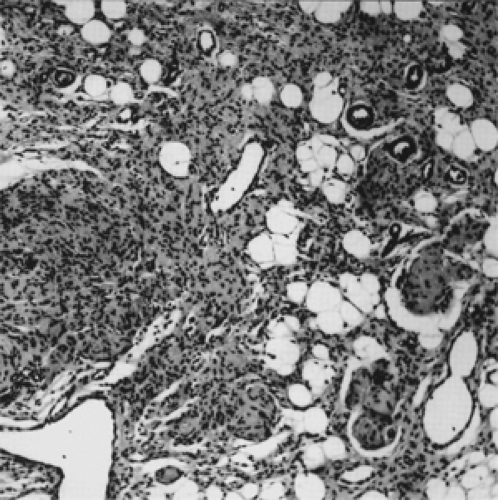 Figure 8.7 Diffuse neurofibroma. Note the nerve tissue permeating orbital fat and surrounding blood vessels (original magnification × 65). |
Neurofibromatosis Type 1
The plexiform neurofibroma is the hallmark of the orbital form of NF1, a peripheral nerve sheath subset of von Recklinghausen neurofibromatosis, first described by von Recklinghausen (1882). The latter is an autosomal dominant abnormality that may involve multiple organ systems. The genetic defect is located on chromosome 17.
Incidence
Our 50-year survey includes 31 pathologically proven plexiform neurofibromas (see Table 3.3). This frequency was approximately three times (31:9) that of neurofibroma and 1½ times more common than schwannoma. The sex incidence was nearly equal, 17 men and 14 women.
The age of the patient when first seen at Mayo Clinic is a meaningless statistic because it does not represent the true age at onset. The age at onset was known in 30 of our patients. The plexiform subset of the tumor is predominantly a disorder with an onset in infancy or childhood. In nearly half our patients (13/30), the tumor was present at birth. Onset at 6 months or earlier occurred in another four patients.
Clinical Features
A thickening or palpable lump in the upper eyelid is the usual sign that brings the infant or child to the attention of a pediatrician or an ophthalmologist. The droopy eyelid may conceal a slight degree of proptosis or displacement of the underlying eye. If further examination of the patient’s body surface reveals several café au lait spots, it is likely the patient will later show other signs or symptoms of generalized NF1. The growth of the tumor is slow. The hypertrophy of the eyelid becomes more localized to its lateral third, and a tumor consisting of cords is palpable. The uneven growth of tumor gives the affected eyelid a sinuous curve. Downward displacement of the eye soon follows owing to extension of the tumor through the orbital septum into the anterosuperior orbit. If the eyelid is particularly thick, it may conceal the subtle malposition of the eye at the time of presentation, unless a simultaneous bulging buphthalmic eye is present. Eventually, by adolescence, the eyelid tumor may further extend into the neighboring temple or the forehead or fill the orbit with a ropy mass likened to a “bag of worms.” With such progression, some mild degree of pain or discomfort may occur because of the localization of orbital neurofibroma in the sensory nerves.
Another pattern of presentation is a proptosed eye associated with pulsation, with or without a palpable mass. The pulsation is best appreciated by viewing the affected eye from its lateral side. The pulsation is caused by incursion of brain tissue into the orbit, secondary to the absence (dysgenesis) of the wings of the sphenoid bone.
Less frequent is the association of optic nerve glioma and the plexiform neurofibroma. Optic nerve glioma is one of several tumors of the central nervous system that present this association. Incidence estimates in the literature range from 10% to 50%. Probably the most accurate survey is the prospective study by Lewis et al. (1984). Their study comprised 217 patients aged 4 weeks to 69 years, and diagnosis was based on stringent criteria.
The incidence of glioma confined to the optic nerves was 10% (22/217). The mean age of this subgroup with radiographic diagnosis of unilateral or multicentric optic nerve glioma was 20.8 years. There was no difference in the sex incidence. Whatever the true incidence of unilateral cases, bilateral optic nerve glioma is considered virtually pathognomic of von Recklinghausen syndrome (Stern et al., 1980).
Several case reports of NF1 have appeared in the literature since our third edition. The first report (Mamalis et al., 1988) was an 8-month-old girl with a tumor that extended from the cavernous sinus into the left orbit involving the optic nerve, extraocular muscles, and eyelid. This case was unusual in that glaucoma developed before clinically evident eyelid manifestations.
The second case (Pittet et al., 1997) was most unusual. This patient, a 15-year-old girl from West Africa, was first seen with a slowly growing gigantic facial tumor since the
age of 5 years (see Fig. 8.8). The mass originated in the right orbital area and completely covered the right side of her face. The pendulous mass extended further inferiorly to the middle of her chest. It measured 35 × 12 cm. The first surgical procedure was excision of the protruding mass. The specimen weighed 2,500 g.
age of 5 years (see Fig. 8.8). The mass originated in the right orbital area and completely covered the right side of her face. The pendulous mass extended further inferiorly to the middle of her chest. It measured 35 × 12 cm. The first surgical procedure was excision of the protruding mass. The specimen weighed 2,500 g.
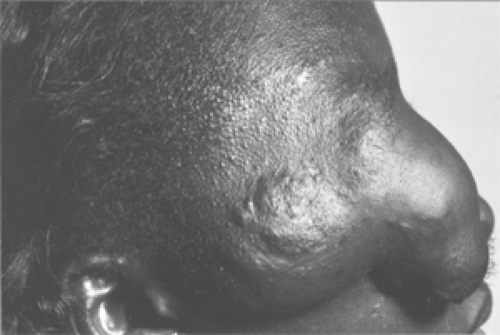 Figure 8.8 Side view of 15-year-old girl showing marked deformity, thickening, and ptosis of right eyelid that completely obscures the right eye. |
The third case (Tada et al., 1998) was a 10-year-old boy with slowly progressive protrusion of the right eye since birth. Exophthalmos measured 15 mm. Imaging studies showed a diffuse, retrobulbar orbital mass that extended into the cavernous sinus through the superior orbital fissure, indicating the tumor was associated with cranial nerves III, IV, V, and VI rather than the optic nerve. Initial diagnosis of NF1 was made because of multiple café au lait spots on the skin of the trunk.
Two of our patients with NF1 are depicted in Figures 8.9 and 8.10.
Hadjistilianou et al. (2002) reported two children with NF1 associated with embryonal rhabdomyosarcoma of the orbit. The first patient was a 4-year-old child who presented with a rapidly progressive proptosis of the left eye. Imaging studies revealed right sphenoid wing agenesis and an orbital mass. At orbitotomy, the mass stained positively for immunostains MyoD1, vimentin, and desmin and negatively for S-100 protein. The second patient was 14 months old with a tumor of the right medial rectus muscle that stained positively for vimentin and desmin and negatively for S-100 protein. The child also had a family history of NF1 and ten café au lait spots on the trunk, which met the criteria for diagnosis of NF1.
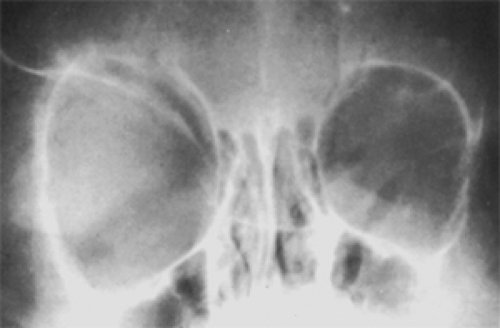 Figure 8.9 Marked enlargement of right orbit in a 4-year-old child. |
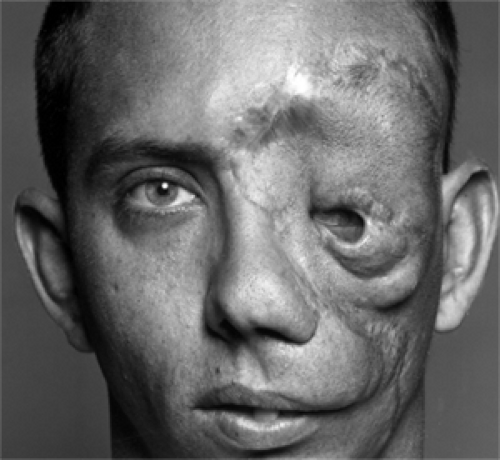 Figure 8.10 This patient with NF1 underwent numerous surgeries to correct deformity of left eyelids to little avail. |
The report by Farris and Grove (1996) described in detail the clinical features associated with NF, their clinical evolution, the myriad associated anomalies, and management of a series of ten patients followed up till the age of 18 years. This publication includes 133 references.
In 1987, the National Institutes of Health proposed strict criteria for the diagnosis of NF. A revision by Mulvihill et al. (1990) defines the diagnostic criteria as two or more of the following:
Six or more café au lait macules >5 mm in greatest diameter in prepubertal patients and >15 mm in greatest diameter in postpubertal patients
Two or more neurofibromas of any type or one plexiform neurofibroma
Freckling in the axillary or inguinal regions
Optic glioma
Two or more Lisch nodules
A distinctive osseous lesion, such as sphenoid dysplasia, or thinning of the long bone cortex with or without pseudarthrosis
A first-degree relative (parent, sibling, or offspring) with NF by the above criteria
Subsequently, Farris and Grove (1996) proposed an eighth addition to the above scheme, namely, a distinct defect on chromosome 17.
Imaging Aspects
When obtaining a good CT scan image may be difficult, particularly in infants and children, skull radiography is still a valued adjunct to diagnosis. In small children with unilateral proptosis and a poorly defined, palpable orbital mass, the skull film may show an enlargement of the bony orbit or, a more positive diagnostic sign, an absence of one or both wings of a sphenoid bone (see Fig. 8.11). In older patients, CT images show all these defects. Figure 8.12 shows the homogeneous, contrast-enhancing character of a multilobular lesion. Magnetic resonance imaging (MRI) demonstrates heterogeneous hypointensity on T1-weighted images relative to orbital fat, but high signal intensity on T2-weighted images relative to orbital fat. The tumor may show a variable degree of contrast enhancement better seen with fat-suppression techniques.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


