Purpose
To review the progress made in understanding the genetic basis, molecular pathology, and treatment of retinoblastoma since the previous Jackson lecture on the topic was published 50 years ago.
Design
Perspective based on personal experience and the literature.
Methods
The literature regarding retinoblastoma was reviewed since 1963. Advances in understanding the biology and treatment of retinoblastoma provided context through the author’s clinical, pathologic, and research experiences.
Results
Retinoblastoma was first identified in the 1500s and defined as a unique clinicopathologic entity in 1809. Until the mid-1900s, knowledge advanced sporadically, with technological developments of ophthalmoscopy and light microscopy, and with the introduction of surgical enucleation, chemotherapy, and radiation therapy. During the last 50 years, research and treatment have progressed at an unprecedented rate owing to innovations in molecular biology and the development of targeted therapies. During this time period, the retinoblastoma gene was discovered; techniques for genetic testing for retinoblastoma were developed; and plaque brachytherapy, chemoreduction, intra-arterial chemotherapy, and intraocular injections of chemotherapeutic agents were successfully introduced.
Conclusions
Nearly all patients with retinoblastoma in developed countries can now be cured of their primary cancer—a remarkable achievement for a childhood cancer that once was uniformly fatal. Much of this success is owed to deciphering the role of the Rb gene, and the benefits of targeted therapies, such as chemoreduction with consolidation as well as intra-arterial and intravitreal chemotherapies. Going forward, the main challenge will be ensuring that access to care is available for all children, particularly those in developing countries.
It is an honor to deliver the LXXI Edward Jackson Memorial Lecture. Although Dr Jackson is probably best known for the Jackson cross cylinder, he profoundly influenced the entire course of modern ophthalmology. Dr Jackson was a major initiator of the predecessor society of the American Academy of Ophthalmology, founder of the American Board of Ophthalmology, and the founding editor of the American Journal of Ophthalmology . In addition to being an educator and clinician, Dr Jackson was known for his grace and kindness, qualities that serve as an example for today’s ophthalmologists.
When Dunphy delivered the XX Jackson Memorial Lecture in 1963, he called his lecture “The Story of Retinoblastoma.” In this lecture, he recounted the history of retinoblastoma, from the 1500s to the mid 1960s. Many advances have been made in the 50 years since then; for instance, the Knudson 2-hit hypothesis, cloning the retinoblastoma gene, chemoreduction therapy, and intra-arterial chemotherapy, to list just a few. Dunphy divided the history of retinoblastoma into 4 general periods: the prehistologic, histologic, enucleation, and irradiation/chemotherapy periods. The retinoblastoma story revolved around numerous personalities, including ophthalmologists, pathologists, and researchers. It is time for an update of that story. I propose that there have been 3 additional periods since Dunphy’s lecture: (1) the period of molecular biology; (2) the period of targeted therapy; and (3) and the period of global health awareness. As in Dunphy’s time, the current era is made possible because of the contributions of ophthalmologists, pathologists, and researchers. Although there have been tremendous advances in understanding the biology and treatment of retinoblastoma, a major challenge persists today: that of ensuring access to care in many parts of the world.
History
In addition to Dunphy’s lecture, Albert has provided an historic review of retinoblastoma. Summarizing briefly, Pawius of Amsterdam is credited as the first to recognize retinoblastoma in an autopsy report of a young child published in 1597. Wardrop of Edinburgh established retinoblastoma as a distinct entity in 1809 and advocated enucleation as the preferred treatment. Steven at the New York Hospital is believed to have reported the first case in the American literature in 1818. In those days, retinoblastoma was known as fungus haematodes , a term coined by Hey of Leeds, England. In 1854, the famous German pathologist, Virchow, introduced the term glioma of the retina . Hirschberg later divided these gliomas into the exophytum and endophytum types, known today as exophytic and endophytic growth patterns. In 1891, Flexner of Johns Hopkins described the histologic finding of cellular rosettes in the tumor ; in 1897, Wintersteiner of Vienna, who was apparently unaware of Flexner’s paper, described the structure’s lumen, a component that now permits subclassification as the Flexner-Wintersteiner rosette. This clear-center rosette is attributable to a recapitulation of the external limiting membrane of the retina. The central portion of the Homer Wright rosette, which can be found in retinoblastoma as well, is filled with neuropil. This feature can be seen in medulloblastoma and neuroblastoma; thus it lacks the specificity that the Flexner-Wintersteiner rosette has for retinoblastoma. Langenbeck and Robin noted in the early 1800s that the tumor microscopically arose in the retina. Histologic resemblance of the tumor to undifferentiated embryonic retina prompted the famous American ophthalmic pathologist, Verhoeff, to name the tumor retinoblastoma , a term that was adopted by the American Ophthalmological Society in 1926. Cases of spontaneously regressed retinoblastoma in phthisical eyes began to appear in the literature in the early 1900s. Gallie and associates described a clinically benign variant of retinoblastoma they called retinoma in 1982. The following year, Margo and associates at the Armed Forces Institute of Pathology reported the histologic features of this benign variant that they called retinocytoma. Trilateral retinoblastoma was described in the early 1980s. This entity includes bilateral retinoblastoma and a third primary intracranial tumor, which may be a primitive neuroectodermal tumor, pineal blastoma, or ependymoblastoma. Another unusual variant of retinoblastoma is anterior diffuse retinoblastoma. This tumor arises in the far peripheral retina as a small, inconspicuous intraretinal mass and seeds the anterior chamber via the aqueous, causing a pseudohypopyon.
Clinical Features
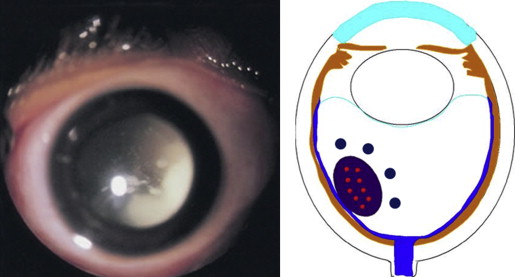
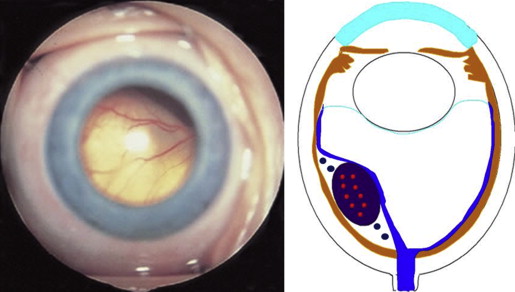
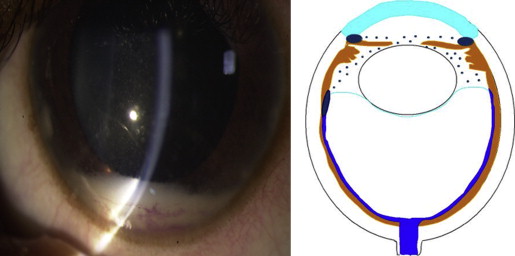
Retinoblastoma, the most common intraocular malignancy in childhood, comprises 4% of all pediatric cancers. The incidence of retinoblastoma is approximately 1 in 20 000 live births and there are about 200–300 new cases of retinoblastoma in the United States each year. In more populated countries, such as India and China, there are about 1000 new cases of retinoblastoma annually. Leukocoria is the most common presenting sign (60%), followed by strabismus (20%). Tumor growth patterns include endophytic ( Figure 1 ), in which the tumor grows into the vitreous; exophytic ( Figure 2 ), in which the tumor grows under the retina toward the choroid; mixed (both endophytic and exophytic); and diffuse infiltrating , which forms a grayish plaque on the retinal surface. A particular variant of the diffuse growth pattern is anterior diffuse ( Figure 3 ). As mentioned above, a small primary tumor arises in the peripheral retina in this variant, then seeds the anterior chamber via the aqueous.
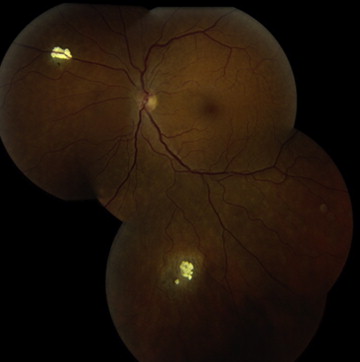
The clinical differential diagnosis of retinoblastoma includes Coats disease, persistent fetal vasculature, vitreous hemorrhage, ocular toxocariasis, familial exudative vitreoretinopathy, rhegmatogenous retinal detachment, coloboma, astrocytic hamartoma, combined hamartoma of the retina/retinal pigment epithelium, and endogenous endophthalmitis ( Table 1 ). The benign variant of retinoblastoma, retinocytoma, may clinically appear as regressed retinoblastoma with associated calcification ( Figure 4 ).
| Coats disease | 40% |
| Persistent fetal vasculature | 28% |
| Vitreous hemorrhage | 5% |
| Ocular toxocariasis | 4% |
| Familial exudative vitreoretinopathy | 3% |
| Rhegmatogenous retinal detachment | 3% |
| Coloboma | 3% |
| Astrocytic hamartoma | 2% |
| Combined hamartoma of retina/retinal pigment epithelium | 2% |
| Endogenous endophthalmitis | 2% |
The Reese-Ellsworth Classification of clinical stage of the disease was published in 1963 and has now been largely supplanted by the International Classification ( Table 2 ), which was developed by a team of retinoblastoma experts in Paris in 2003, including Murphree, Desjardins, Gallie, Shields, Shields, and others. Retinoblastoma spreads by 3 pathways: anteriorly via the vitreous/subretinal space into the anterior segment; posteriorly via the optic nerve into the cranial cavity; and externally via the choroid to the systemic circulation or through the sclera into the orbit. Prognostic risk factors for dissemination include optic nerve and/or orbital invasion and massive choroidal invasion.
| Group | Subgroup | General | Specific Features |
|---|---|---|---|
| A | A | Small tumor | Retinoblastoma ≤3 mm in size |
| B | B | Larger tumor | Retinoblstoma >3 mm in size or |
| Macula | Macular retinoblastoma ≤3 mm to fovea | ||
| Juxtapapillary | Juxtapapillary retinoblastoma location ≤1.5 mm to disc | ||
| Subretinal fluid | Clear subretinal fluid ≤3 mm from margin | ||
| Focal seeds | Retinoblastoma with | ||
| C | C1 | Subretinal seeds ≤3 mm from retinoblastoma | |
| C2 | Vitreous seeds ≤3 mm from retinoblastoma | ||
| C3 | Both subretinal and vitreous seeds ≤3 mm from | ||
| Diffuse seeds | Retinoblastoma with | ||
| D | D1 | Subretinal seeds >3 mm from retinoblastoma | |
| D2 | Vitreous seeds >3 mm from retinoblastoma | ||
| D3 | Both subretinal and vitreous seeds >3 mm from | ||
| E | E | Extensive retinoblastoma | Extensive retinoblastoma occupying >50% globe or |
| Neovascular glaucoma | |||
| Opaque media from hemorrhage in AC, vitreous, or SR space | |||
| Invasion of postlaminar optic nerve, choroid, sclera, orbit, AC |
Second primary tumors in hereditary retinoblastoma survivors represent a greater risk of death than the retinoblastoma itself. The cumulative risk for these tumors is roughly 1% per year, reaching 50% at 50 years. Radiotherapy, especially to the site of retinoblastoma before age 1, greatly increases the chances of a second tumor. These tumors are mainly sarcomas, in particular leiomyosarcoma in the field of radiation and osteosarcoma outside of the field.
Clinical Features
Retinoblastoma, the most common intraocular malignancy in childhood, comprises 4% of all pediatric cancers. The incidence of retinoblastoma is approximately 1 in 20 000 live births and there are about 200–300 new cases of retinoblastoma in the United States each year. In more populated countries, such as India and China, there are about 1000 new cases of retinoblastoma annually. Leukocoria is the most common presenting sign (60%), followed by strabismus (20%). Tumor growth patterns include endophytic ( Figure 1 ), in which the tumor grows into the vitreous; exophytic ( Figure 2 ), in which the tumor grows under the retina toward the choroid; mixed (both endophytic and exophytic); and diffuse infiltrating , which forms a grayish plaque on the retinal surface. A particular variant of the diffuse growth pattern is anterior diffuse ( Figure 3 ). As mentioned above, a small primary tumor arises in the peripheral retina in this variant, then seeds the anterior chamber via the aqueous.
The clinical differential diagnosis of retinoblastoma includes Coats disease, persistent fetal vasculature, vitreous hemorrhage, ocular toxocariasis, familial exudative vitreoretinopathy, rhegmatogenous retinal detachment, coloboma, astrocytic hamartoma, combined hamartoma of the retina/retinal pigment epithelium, and endogenous endophthalmitis ( Table 1 ). The benign variant of retinoblastoma, retinocytoma, may clinically appear as regressed retinoblastoma with associated calcification ( Figure 4 ).
| Coats disease | 40% |
| Persistent fetal vasculature | 28% |
| Vitreous hemorrhage | 5% |
| Ocular toxocariasis | 4% |
| Familial exudative vitreoretinopathy | 3% |
| Rhegmatogenous retinal detachment | 3% |
| Coloboma | 3% |
| Astrocytic hamartoma | 2% |
| Combined hamartoma of retina/retinal pigment epithelium | 2% |
| Endogenous endophthalmitis | 2% |
The Reese-Ellsworth Classification of clinical stage of the disease was published in 1963 and has now been largely supplanted by the International Classification ( Table 2 ), which was developed by a team of retinoblastoma experts in Paris in 2003, including Murphree, Desjardins, Gallie, Shields, Shields, and others. Retinoblastoma spreads by 3 pathways: anteriorly via the vitreous/subretinal space into the anterior segment; posteriorly via the optic nerve into the cranial cavity; and externally via the choroid to the systemic circulation or through the sclera into the orbit. Prognostic risk factors for dissemination include optic nerve and/or orbital invasion and massive choroidal invasion.
| Group | Subgroup | General | Specific Features |
|---|---|---|---|
| A | A | Small tumor | Retinoblastoma ≤3 mm in size |
| B | B | Larger tumor | Retinoblstoma >3 mm in size or |
| Macula | Macular retinoblastoma ≤3 mm to fovea | ||
| Juxtapapillary | Juxtapapillary retinoblastoma location ≤1.5 mm to disc | ||
| Subretinal fluid | Clear subretinal fluid ≤3 mm from margin | ||
| Focal seeds | Retinoblastoma with | ||
| C | C1 | Subretinal seeds ≤3 mm from retinoblastoma | |
| C2 | Vitreous seeds ≤3 mm from retinoblastoma | ||
| C3 | Both subretinal and vitreous seeds ≤3 mm from | ||
| Diffuse seeds | Retinoblastoma with | ||
| D | D1 | Subretinal seeds >3 mm from retinoblastoma | |
| D2 | Vitreous seeds >3 mm from retinoblastoma | ||
| D3 | Both subretinal and vitreous seeds >3 mm from | ||
| E | E | Extensive retinoblastoma | Extensive retinoblastoma occupying >50% globe or |
| Neovascular glaucoma | |||
| Opaque media from hemorrhage in AC, vitreous, or SR space | |||
| Invasion of postlaminar optic nerve, choroid, sclera, orbit, AC |
Second primary tumors in hereditary retinoblastoma survivors represent a greater risk of death than the retinoblastoma itself. The cumulative risk for these tumors is roughly 1% per year, reaching 50% at 50 years. Radiotherapy, especially to the site of retinoblastoma before age 1, greatly increases the chances of a second tumor. These tumors are mainly sarcomas, in particular leiomyosarcoma in the field of radiation and osteosarcoma outside of the field.
Pathology
Retinoblastoma is derived from cells of neuroectodermal origin from the inner layer of the optic cup. The tumor may grow toward the vitreous and/or subretinal space, forming a multilobulated white mass with associated vitreous or subretinal seeds. In these cases, the tumor grows in sleeves and cuffs around central vessels from which it derives its nutrition. When the tumor outgrows its vascular supply 90–110 μm away from the central vascular channel, it undergoes ischemic necrosis and may contain areas of dystrophic calcification, thus yielding a grossly visible chalky or cottage cheese appearance. The tumor may also grow diffusely within the retina and derive its nutrition from the retinal vasculature. Vitreous and subretinal seeds of retinoblastoma are avascular and contain areas of necrosis in the center of the seed away from the vitreous or subretinal fluid. Cytologically, retinoblastoma is made up of small, round, blue cells. Retinoblastoma may exhibit differentiation, which becomes less evident with age. Signs of differentiation, going from least to most differentiated, include Homer Wright or Flexner-Wintersteiner rosettes and fleurettes ( Figure 5 ). Fleurettes are particularly abundant in retinocytoma, which also contain areas of calcification. The 2 most important histologic prognostic factors for distant metastases are optic nerve invasion beyond the lamina cribrosa and “massive” choroidal invasion measuring 3 mm or greater in diameter in which the tumor comes in contact with the sclera. The approximate survival rates for children with retinoblastoma extending into the prelaminar, laminar, and postlaminar optic nerve and surgical margin of resection are >90%, 85%, 60%, and 35%, respectively. The overall survival rate when there is massive choroidal invasion is approximately 70%.
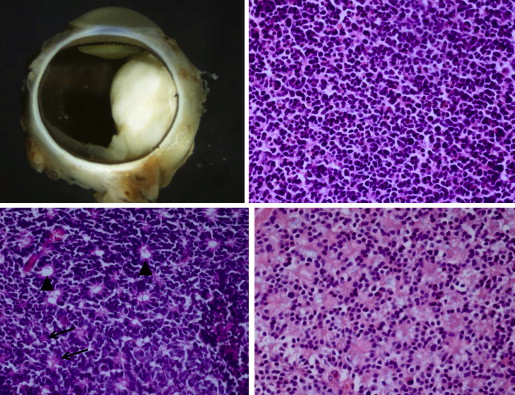
The International Retinoblastoma Staging Working Group (IRSWG) has provided guidelines for handling eyes enucleated for retinoblastoma, including preparation of the pupil–optic nerve section and breadloafing the calottes to evaluate for choroidal invasion. It is estimated that approximately 20% of eyes enucleated with retinoblastoma in the United States will exhibit high-risk features. Clinical predictors of high-risk histologic features include older patient age, time from diagnosis to enucleation, hyphema, pseudohypopyon, staphyloma, orbital cellulitis, and elevated intraocular pressure. Histologic examination of enucleated eyes after chemoreduction therapy for retinoblastoma may show type 1 (cottage cheese), type 2 (fish flesh), type 3 (combination of types 1 and 2), or type 4 (complete) regression, or histologically intact, presumably viable tumor cells. The type 2 regression pattern may result in retinocytoma-like clinical appearance; the tumor may have the same histologic features. It is not surprising that regressed tumors have the same histologic appearance of retinocytoma, since well-differentiated tumors are presumably relatively resistant to chemotherapy, as cells are not cycling. Tumors treated by intra-arterial chemotherapy (IAC) exhibit similar regression patterns. Treatment failures are attributable to persistence of vitreous seeds, subretinal seeds, intraretinal tumor, or tumor that is unresponsive to chemotherapy.
Molecular Biology
The information in this section represents our current understanding of the molecular mechanisms and pathways involved in development, extraocular extension, and metastasis of retinoblastoma based on evidence available at this time; some of this information will likely prove to be incorrect or an oversimplification of these mechanisms. The understanding of these mechanisms will change as experimental investigations will further clarify these molecular pathways in the future.
Genetics and Molecular Mechanisms
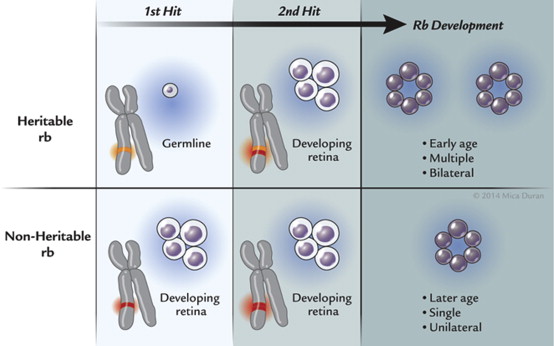
The most far-reaching advances in retinoblastoma since Dunphy’s lecture have been in understanding the genetics and molecular origins of the tumor and mechanisms of progression. These fields of endeavor have provided major breakthroughs in deciphering the pathogenesis of cancer. It had long been known that retinoblastoma could be sporadic or heritable when Knudson at the MD Anderson Cancer Center developed his now celebrated “2-hit” hypothesis. Knudson had graphed the percent of undiagnosed cases vs age at diagnosis and found 2 distinct curves, 1 linear and the other exponential. The linear curve corresponded to patients who had bilateral tumors and theoretically inherited 1 copy of a mutated gene (germline mutation) and, as their retina developed, acquired the second mutation (second “hit”). The exponential curve corresponded to children who had unilateral tumors and acquired both mutations in their developing retina. The heritable variety of retinoblastoma may be explained by the inheritance of a germline mutation in combination with an acquired mutation in the developing retina, whereas the acquired variety of retinoblastoma develops after mutations in both alleles of the retinoblastoma gene arise in the developing retina ( Figure 6 ). Individuals who harbored a germline mutation in all cells were predisposed to develop a variety of other malignancies. As the field progressed and laboratory techniques improved, investigators were able to localize the retinoblastoma gene (RB1) to chromosome 13q14.2. In 1984, Dryja’s laboratory cloned the RB1 gene, a landmark achievement as this was the first human tumor suppressor gene cloned. The gene was subsequently sequenced. The RB1 gene was found to be large (approximately 200 kb) and had 27 dispersed exons. Two structurally related retinoblastoma-like genes (RBL1, RBL2) have since been identified. RBL1 and RBL2 are located at chromosomes 20q11.2 and 16q12.2, respectively. Investigators have recently found that retinoblastoma may arise after MYC mutation in the absence of RB mutations. These tumors appear to be diagnosed at an earlier age than those with RB mutations. It became evident that although mutations of both alleles of the RB1 gene were necessary in most cases, they were not sufficient for phenotypic expression. Dimaras and associates found that loss of both copies of RB1 may lead to retinoma (retinocytoma) and progressive genomic instability in retinoma then leads to retinoblastoma. Gallie’s laboratory found that additional mutational events (“3 hits”) were required for progression to a fully a malignant tumor.
The function of RB has been described by Harbour. The protein (Rb), composed of 928 amino acids, is a nuclear phosphoprotein that contains an N-terminal region, a central pocket domain with A and B boxes, and a C terminus. The pocket is formed by interactions of the A and B boxes and tumor-forming mutations disrupt this pocket. Rb binds to the chromatin remodeling proteins HDAC and BRG1 via the LxCxE binding site and to E2F transcription factors via its E2F binding site. Rb is multifunctional: it suppresses transcription via histone deacetylases (HDACs) and BRG; it also regulates the cell cycle via E2F. Phosphorylated Rb binds directly to E2F and blocks the E2F transactivation domain. When Rb is dephosphorylated via p16, chromatin remodeling proteins (HDAC, BRG1), which bind to Rb, alter chromatin structure and prevent access to transcription, thus leading to exit from the cell cycle. When Rb is phosphorylated, HDAC and BRG1 are released, thus allowing progression through the cell cycle by de-repression of cyclins owing to sequential Rb phosphorylation involving cyclins and cyclin-dependent kinases (CDKs). Under stress and excess proliferation, phosphorylation of Ser567 changes the tertiary structure of Rb, releasing E2Fs and leading to apoptosis. Thus, Rb plays a central role in the fate of a cell through either exit of the cell cycle or apoptosis. Mutations in Rb and/or inhibition of Rb by oncoproteins produced by tumor-causing viruses (SV40, adenovirus, human papillomavirus) abrogate Rb’s ability to mediate exit from the cell cycle or apoptosis and lead to uncontrolled cell division, a hallmark of cancer. Perturbations in genes such as BRCA2 on chromosome 13, CDKN2 on chromosome 9, and TP53/BRCA1 on chromosome 17 cooperate with alterations in Rb by promoting tumor progression. Breast cancer–related genes BRCA1 and BRCA2 are involved with DNA damage-response pathways and, in addition to viral oncoproteins, may directly alter the function of Rb. Loss of p16/INK or p16/INK promoter hypermethylation may alter cell cycle exit. Additionally, alterations in the phosphorylated retinoblastoma protein (pRb) phosphorylation/inactivation by CDK4/cyclin D1 dysregulates Rb’s ability to promote cell cycle exit and TP53 loss facilitates cell survival. These other events, or “third hits,” appear to be necessary for human retinoblastoma to progress. RB1/pRb inactivation results in loss of its ability to regulate exit from the cell cycle and upregulates ZEB protein, a transcriptional repressor of E-cadherin, a determinant of epithelial to mesenchymal transformation that allows for transmigration and invasion, 2 other hallmarks of cancer. The biologic events that lead to retinoblastoma formation are summarized in Figure 7 .



