Purpose
To evaluate the cellular nature of and diagnostic terminology used in connection with acquired retinal “vasoproliferative tumors.”
Design
Retrospective clinicopathologic study.
Methods
Clinical records and microscopic slides of 4 enucleated globes were reviewed. Special stains and immunohistochemical probes for CD31, CD34, p53, glial fibrillary acidic protein (GFAP), CD163, and Ki67 (cell replication) were employed; ultrastructural and fluorescence in situ hybridization (FISH) analyses were performed.
Results
Tumors were located inferotemporally in middle-aged patients. They were uniformly composed of compacted elongated, GFAP-positive spindle cells (due to intermediate filaments identified ultrastructurally) with a Ki67 index of less than 1%. Rosenthal fibers and eosinophilic granular bodies were observed. Hyalinized periodic acid–Schiff-positive vessels were widely separated. CD31 and CD34 revealed a sparse microvasculature. Tumor-associated exudate spread predominantly subretinally. The retinal pigment epithelium had undergone extensive placoid fibrous metaplasia with focal ossification. P53 upregulation, BRAF-KIAA gene rearrangement, and IDH1 R132H mutation typically associated with low-grade astrocytic neoplasms were absent.
Conclusions
Retinal “vasoproliferative” tumors have been mischaracterized, because they actually display a paucity of microvessels. Proliferating fibrous astrocytes with a very low proliferation index predominate, without immunohistochemical or genetic evidence favoring a neoplasm. Subretinal exudate appeared capable of provoking widespread fibrous metaplasia of the pigment epithelium that was mainly responsible for secondary retinal damage. The term “reactive retinal astrocytic tumor” is proposed as more appropriate for this entity. In carefully selected progressive lesions, consideration should be given to earlier surgical intervention before extensive subretinal exudate accumulates and pigment epithelial proliferation with fibrous metaplasia ensues.
Acquired retinal tumors containing both vascular and glial components were first described by Shields and associates in 1983 in a series of 12 cases. Their initial term for this condition was “acquired retinal hemangioma,” based on the fluorescein angiographic features. Subsequently, the same ocular oncology service reported a series of 103 patients with 129 tumors in 1995; at that time, the authors rechristened the lesions “vasoproliferative tumors of the retina” (VPTR). Many other synonyms have thereafter been employed—“acquired retinal angioma,” “angioma-like mass” in retinopathy of prematurity, “retinal angiomatous mass” after retinal detachment surgery, “retinal angiomas in the aged,” “angioma-like lesion” in sickle cell disease, “glioangiosis,” “presumed acquired retinal hemangioma,” “hemangioma-like masses of the retina,” “peripheral retinal telangiectasia” simulating melanoma, “peripheral uveal neovascularization,” and “neovascular fundus abnormality” in patients with uveitis —all of which share an emphasis on vasogenesis as the essential pathologic substrate. There are, however, few studies on the histopathologic features of these lesions upon which to judge their true nature. We have been able to evaluate 4 globes harboring these tumors with in-depth histopathologic, immunohistochemical, ultrastructural, and genetic studies. Specifically, our first objective is to determine whether vasoproliferation or progressive astrocytic proliferation drives the condition. Secondly, if the astrocytes are basically responsible for the formation of the tumors and their progressive enlargement, are they reactive or neoplastic?
Methods
This study was performed under the auspices of the Massachusetts Eye and Ear Infirmary Institutional Review Board (IRB): Examination of Archived Ocular Tissues for Research Studies on Ocular Structure, Function, Physiology and Association with Various Ocular Diseases, 196320-3; and the Emory Eye Center, Emory University School of Medicine IRB: Massive Retinal Gliosis, 00052749; and was conducted in compliance with the rules and regulations of the Health Insurance Portability and Accountability Act and in adherence to the Declaration of Helsinki and all federal and state laws. The files of the David G. Cogan Laboratory of Ophthalmic Pathology at the Massachusetts Eye and Ear Infirmary, and of the L.F. Montgomery Eye Pathology Laboratory at the Emory Eye Center, were retrospectively reviewed for the period 1997-2012 for cases diagnosed as retinal tumor, vasoproliferative tumor, astrocytoma, or astrocytic scar. Two cases at each institution were identified for inclusion in this study after critical review had been independently performed and agreed upon by 2 of the authors (H.E.G. and F.A.J.). Additionally, 2 neuropathologists (an author of this article [D.J.B.] and Dr Tessa Hedley-Whyte of the Neuropathology Division of the Massachusetts General Hospital) also studied the microscopic slides. Hospital records, fundus photographs, laboratory reports of fluorescein angiograms and ultrasonograms, and office files documenting patient examinations were retrieved when available and clinical information was extracted. For comparison purposes, an enucleated globe with a retinal hemangioblastoma (von Hippel lesion) was studied with the same pathologic and immunohistochemical methods mentioned below, excluding the genetic investigations.
Paraffin-embedded microscopic sections prepared from the enucleated eyeballs were stained with hematoxylin-eosin, periodic acid–Schiff (PAS), and Masson trichrome stains. Immunohistochemical staining was conducted using the following probes: glial fibrillary acidic protein (GFAP) for glial cell intermediate cytoplasmic filaments; S-100 for Schwann cells; CD31 and CD34 for vascular endothelium; alpha smooth muscle actin and calponin for smooth muscle differentiation; CD163 for histiocytes; synaptophysin for evidence of synaptogenesis indicating neuronal differentiation; neurofilament for neuronal dendrites and axons; p53 for upregulation in astrocytomas; and Ki67 for cells in S-phase of DNA replication prior to mitosis. Portions of 1 tumor were fixed in 2% glutaraldehyde, processed, and transmission electron microscopy was performed (JEOL 100CX; JEOL, Tokyo, Japan). Fluorescence in situ hybridization (FISH) for the fusion gene product of KIAA1549 and BRAF was performed on 5-μm-thick paraffin-embedded tissue sections, as described elsewhere. The methodological characterization of IDH1 , R132H, and the application of p53 to astrocytomas have also been previously described. Briefly, for the astrocytic gene studies the probes for KIAA and BRAF included a fluorescein isothiocyanate (FITC)-labeled locus-specific probe RP11-355D18 (CHORI BACPAC Resources Center, Oakland, California, USA) corresponding to FITC-labeled KIAA1549 (green) and a rhodamine-labeled locus-specific probe 726N20 corresponding to BRAF (red). Brain biopsy specimens were used as negative controls. Tumors were scored as positive for the BRAF-KIAA1549 fusion when >25% of the cells demonstrated yellow signals (indicating overlap of the green and red signals), along with associated copy-number gains (at least 3 green and/or red signals) in at least 100 nonoverlapping, intact nuclei. Pyrosequencing for the activating BRAF V600E mutation was performed using a PyroMark W96d (Qiagen, Valencia, California, USA). A portion of the gene including codon 600 was amplified by polymerase chain reaction and then sequenced across codon 600.
Results
Clinical Findings
In the present group of cases, 3 men, aged 36, 70, and 75 years, and 1 woman, aged 32 years, were affected. Each patient gave a history of either uveitis, retinal detachment surgery with scleral buckling, ocular trauma (up to 30 years earlier), or penetrating keratoplasty. The reason for enucleation was a blind, painful eye. Prior to enucleation, 2 patients were known to have an intraocular tumor; 1 of these at one time had had vision of 20/400 with an earlier diagnosis of amblyopia in the involved eye. The following brief case report exemplifies the diagnostic and therapeutic aspects of these lesions.
Case Description
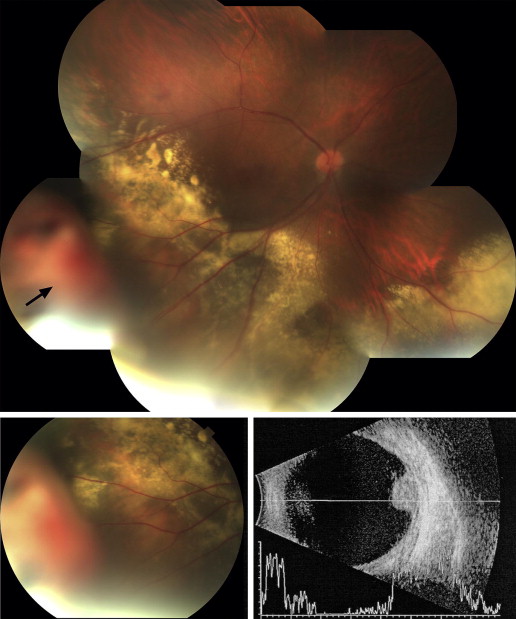
A 36-year-old man was referred to a retina specialist by his general ophthalmologist for evaluation of a retinal mass associated with exudates. He complained of “waves” and a cloudy “blob” in his right eye that he had observed for a month. There was a history of uveitis in both eyes that was first diagnosed at age 5, in addition to amblyopia in the right eye. The first visual acuities obtained by the specialist were 20/400 OD and 20/25-3 OS. Intraocular pressure was 18 mm Hg in both eyes. There was mild nuclear sclerosis and a mild posterior subcapsular cataract in the right eye, and a mild posterior subcapsular cataract in the left eye. The remainder of the anterior segment examination was unremarkable. The retinal examination of the right eye ( Figure 1 ) revealed a pink-orange, elevated, and polypoidal lesion that was approximately 5 mm in height and 10 mm in basal diameter in the right inferotemporal far periphery. There was a surrounding shallow nonbullous retinal detachment with an exudative response that extended toward the macula and into the inferonasal periphery. There were no intraretinal telangiectasias. Retinal examination of the left eye revealed pigment epithelial dropout inferior to the center of the macula and in the inferior and nasal midperiphery as well as in the far periphery. Intravenous fluorescein angiography demonstrated early hyperfluorescence and late leakage of the lesion. Two weeks after presentation, the patient underwent cryotherapy to the base of the lesion and laser treatment to the surrounding shallow exudative retinal detachment. Although he initially reported an improvement in his visual symptoms, he subsequently developed refractory neovascular glaucoma over 16 months. After an unavailing course of intravitreal injections of bevacizumab (1.5 mg, 4 injections), the patient underwent enucleation for a blind, painful eye.
Histopathologic and Immunohistochemical Findings
The 4 enucleated globes measured between 25 × 26 × 24 mm and 20 × 24 × 18 mm. On opening the globes, the vitreous was generally cloudy and a far peripheral retinal tumor was discovered. The tumors measured at their bases and greatest heights 3.0/1.5 mm, 6.0/3.0 mm, 8.0/3.0 mm, and 5.0/7.0 mm. Low-power photomicrographs of 2 tumors illustrate their overall configuration and typical location ( Figure 2 , Top left and Top right). Only 1 of the lesions showed a focus of significant noninflamed bland necrosis toward its apex ( Figure 2 , Top right and Middle left). A rim of viable tumor was present at the very apex of this lesion bordering the vitreous cavity. There was an abrupt interface between the mummified tumor focus with its preserved, noncellular vascular outlines and the underlying viable tumor ( Figure 2 , Middle left). The vessels within the viable tumor and abutting the necrotic area were massively hyalinized, with very narrow lumens.
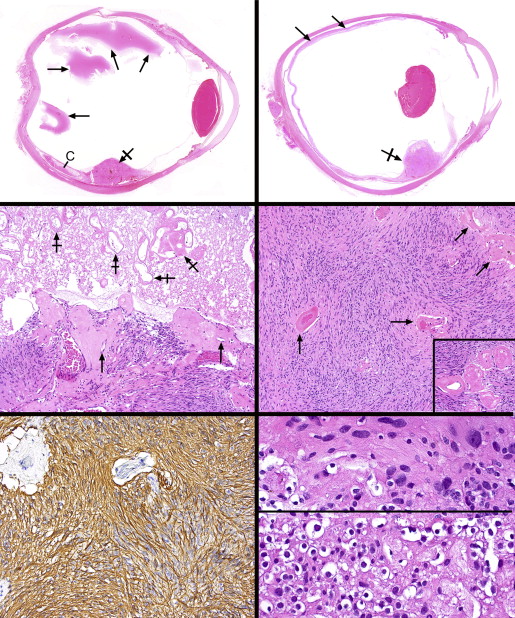
Each tumor was circumscribed but not clearly demarcated from the surrounding atrophic, mildly gliotic, and degenerate retina, with which it blended imperceptibly. The overall architecture was characterized by interweaving bundles of spindle cells and widely separated vascular channels with hyalinized walls ( Figure 2 , Middle right and inset). Intratumoral hemorrhage was not observed, nor were there collections of eosinophilic exudate, pigment-bearing cells, calcospherites, or dystrophic calcification. Antibodies for glial fibrillary acidic protein intensely and positively immunostained all lesions and disclosed very elongated cellular processes resulting in hair-like (pilocytic) bipolar cells ( Figure 2 , Bottom left). Smooth muscle actin and calponin were negative in the tumor cells but immunostained some of the vascular mural cells. Rare, mildly atypical hyperchromatic nuclei were discovered amidst the overwhelmingly bland, oval and elongated tumor cell nuclei ( Figure 2 , Bottom right, top panel). Numerous CD163-positive histiocytes were scattered within the tumors but had not undergone xanthomatization; they were especially prominent in the tumor that had experienced partial necrosis.
Whereas the majority of the tumors was composed of spindle cells, in the smallest lesion a small focus of honeycombed cells with clear cytoplasm and round nuclei resembling oligodendroglial cells was detected at the posterior peripheral edge of the lesion ( Figure 2 , Bottom right, bottom panel). Synaptophysin and neurofilament positivity was discovered among these cells, implying that they were residual retinal elements. Eosinophilic structures within the cytoplasm of some of the tumor cells were noted (Rosenthal fibers) ( Figure 3 , Top left), and collections of variably sized eosinophilic granular bodies were also scattered about ( Figure 3 , Top right). Ki67 immunoreacted with fewer than 1% of nuclei ( Figure 3 , Middle left).
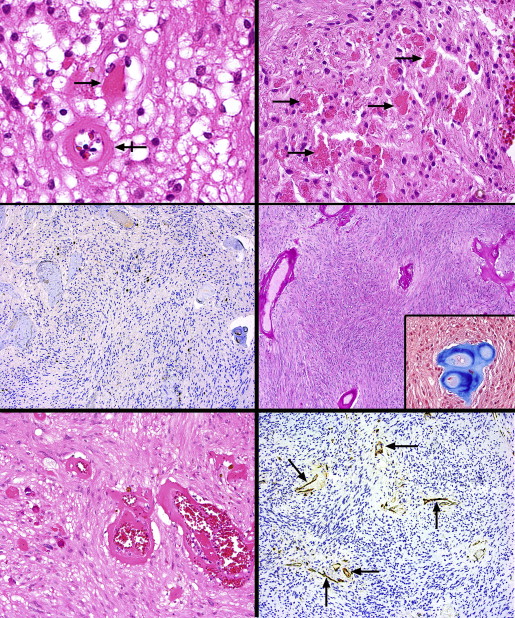
The walls of the hyalinized vessels were PAS-positive and Masson trichrome–positive ( Figure 3 , Middle right and inset). Small vessel proliferation was inconspicuous, but there were occasional clusters with moderate-sized lumens evincing incipiently thickened walls ( Figure 3 , Bottom left). CD31 and CD34 demonstrated a paucity of microvessels that were widely separated and lacked thickened walls ( Figure 3 , Bottom right). For purposes of comparison, a hemangioblastoma of the retina (von Hippel lesion) was also evaluated with CD31 and displayed a luxuriant network of capillary-sized panels, in stark contrast to their scarcity in the current tumors ( Figure 4 , Top left, left and right panels).
One of the more dramatic features associated with the lesions was the extent of retinal pigment epithelial proliferation, which included extensive fibrous and even osseous metaplasia ( Figure 4 , Top right). These phenomena were predominantly noticeable beneath and at the edges of the lesions; in 2 cases a placoid mass of fibrous metaplasia extended throughout the entire circumference of the subretinal space, almost constituting an extra tunic of the eye ( Figure 4 , Middle left). A narrow band of chronic inflammation was frequently detected at the base of the placoid metaplastic changes in the choroid. The retina overlying this placoid fibrous proliferation was moderately gliotic and generally did not display cystic degeneration (except for 1 microcyst with eosinophilic material immediately posterior to 1 tumor), significant intraretinal exudation, or telangiectasia.
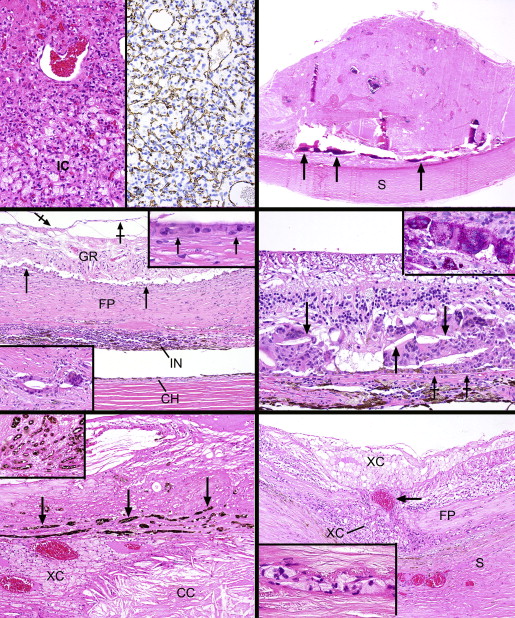
Although there were no bullous retinal detachments with subretinal exudate, a thin lamina of exudate was discovered beneath 1 tumor. The retinas were otherwise mildly separated from the aforementioned fibrous metaplasia of the pigment epithelium. A preretinal gliotic membrane was detected focally in 3 of the 4 globes ( Figure 4 , Middle left). In the space between the retina and the fibrous plaque were collections of epithelioid mononucleated and multinucleated histiocytes ( Figure 4 , Middle left and top inset); sometimes these elements became embedded in the middle of the plaque ( Figure 4 , Middle left, bottom inset). The granulomatous response frequently engulfed cholesterol clefts ( Figure 4 , Middle right). The cytoplasms of the histiocytes were intensely PAS-positive ( Figure 4 , Middle right, inset). Progressively depigmenting pigment epithelial cells gradually blended into the collections of PAS-positive histiocytes ( Figure 4 , Middle right).
In 2 cases the choroid was moderately inflamed (1 focally, the other diffusely) with a lymphoplasmacytic infiltrate; in the others, the uvea was more lightly infiltrated ( Figure 4 , Middle left). An exceptional feature observed once was massive exudate of a sufficient degree to involve the choroid with collections of cholesterol clefts and xanthoma cells ( Figure 4 , Bottom left). In this zone there was an overlying metaplastic pigment epithelial fibrous plaque with pseudoadenomatous units ( Figure 4 , Bottom left and inset). Xanthoma cells were also found in this case in the retina more posteriorly, where a break in the fibrous subretinal plaque allowed a choroidal vessel to contact the outer retina ( Figure 4 , Bottom right). Small collections of xanthoma cells also resided between the lamellae of the sclera ( Figure 4 , Bottom right) and in the optic nerve head. These findings were present in the globe with the most intense vitreous exudate, illustrated in Figure 2 , Top left. All lesions showed cataractous changes of the lens, peripheral anterior synechiae, and iris neovascularization. Anterior segment descemetization (new Descemet membrane deposited beneath the spread of corneal endothelial cells across the chamber angle onto the iris surface) was discovered in 1 globe. An episcleral fibrous capsule of an encircling element was observed in the case that had prior retinal detachment surgery; in another globe there was evidence of a healed posterior scleral wound.
Ultrastructural and Genetic Findings
Electron microscopic evaluation of the tumor cells in 1 case disclosed spindle shapes and myriad smaller interweaving processes ( Figure 5 , Top). The plasmalemmas were straight and there were no intercellular desmosomes, interdigitations, imbrications, or basement membrane deposition. The cytoplasm was endowed with numerous intermediate filaments, a few mitochondria, and short profiles of rough-surfaced endoplasmic reticulum, particularly in the perikaryon. Lysosomes were discovered in the smaller cellular processes. No smooth-surfaced endoplasmic reticulum was observed. The nuclei had clumped heterochromatin and small nucleoli with tightly wound nucleolonemas.
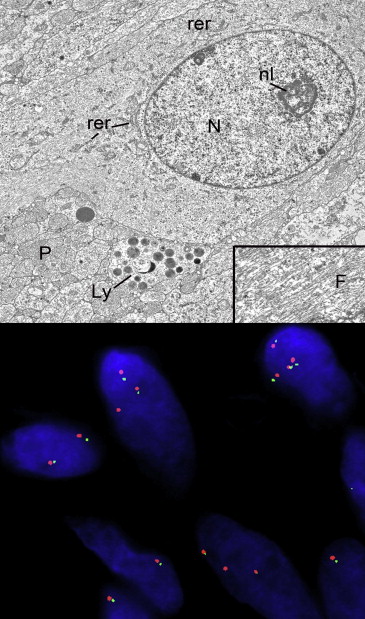
Immunohistochemical staining for the most frequent form of isocitrate dehydrogenase-1 ( IDH1 ) mutant protein, corresponding to the R132H mutation and noted in 70%-80% of infiltrating forms of low-grade glial neoplasms, did not demonstrate immunoreactivity in any of the cases. Immunohistochemistry for p53, which is commonly overexpressed in low-grade infiltrative astrocytomas, was also negative in all cases. FISH for the KIAA-BRAF fusion event, which is noted in a high percentage of cerebellar pilocytic astrocytomas, was performed in 4 cases and uncovered no fusion events in the tumors ( Figure 5 , Bottom). Polysomy 7 was found but was regarded as a nonspecific abnormality that does not indicate a neoplastic condition. Pyrosequencing of the BRAF gene for the activating V600E mutation, a frequent mutation in low-grade gliomas of various histopathologies, revealed no mutations in any of the 4 cases tested.
Results
Clinical Findings
In the present group of cases, 3 men, aged 36, 70, and 75 years, and 1 woman, aged 32 years, were affected. Each patient gave a history of either uveitis, retinal detachment surgery with scleral buckling, ocular trauma (up to 30 years earlier), or penetrating keratoplasty. The reason for enucleation was a blind, painful eye. Prior to enucleation, 2 patients were known to have an intraocular tumor; 1 of these at one time had had vision of 20/400 with an earlier diagnosis of amblyopia in the involved eye. The following brief case report exemplifies the diagnostic and therapeutic aspects of these lesions.
Case Description
A 36-year-old man was referred to a retina specialist by his general ophthalmologist for evaluation of a retinal mass associated with exudates. He complained of “waves” and a cloudy “blob” in his right eye that he had observed for a month. There was a history of uveitis in both eyes that was first diagnosed at age 5, in addition to amblyopia in the right eye. The first visual acuities obtained by the specialist were 20/400 OD and 20/25-3 OS. Intraocular pressure was 18 mm Hg in both eyes. There was mild nuclear sclerosis and a mild posterior subcapsular cataract in the right eye, and a mild posterior subcapsular cataract in the left eye. The remainder of the anterior segment examination was unremarkable. The retinal examination of the right eye ( Figure 1 ) revealed a pink-orange, elevated, and polypoidal lesion that was approximately 5 mm in height and 10 mm in basal diameter in the right inferotemporal far periphery. There was a surrounding shallow nonbullous retinal detachment with an exudative response that extended toward the macula and into the inferonasal periphery. There were no intraretinal telangiectasias. Retinal examination of the left eye revealed pigment epithelial dropout inferior to the center of the macula and in the inferior and nasal midperiphery as well as in the far periphery. Intravenous fluorescein angiography demonstrated early hyperfluorescence and late leakage of the lesion. Two weeks after presentation, the patient underwent cryotherapy to the base of the lesion and laser treatment to the surrounding shallow exudative retinal detachment. Although he initially reported an improvement in his visual symptoms, he subsequently developed refractory neovascular glaucoma over 16 months. After an unavailing course of intravitreal injections of bevacizumab (1.5 mg, 4 injections), the patient underwent enucleation for a blind, painful eye.
Histopathologic and Immunohistochemical Findings
The 4 enucleated globes measured between 25 × 26 × 24 mm and 20 × 24 × 18 mm. On opening the globes, the vitreous was generally cloudy and a far peripheral retinal tumor was discovered. The tumors measured at their bases and greatest heights 3.0/1.5 mm, 6.0/3.0 mm, 8.0/3.0 mm, and 5.0/7.0 mm. Low-power photomicrographs of 2 tumors illustrate their overall configuration and typical location ( Figure 2 , Top left and Top right). Only 1 of the lesions showed a focus of significant noninflamed bland necrosis toward its apex ( Figure 2 , Top right and Middle left). A rim of viable tumor was present at the very apex of this lesion bordering the vitreous cavity. There was an abrupt interface between the mummified tumor focus with its preserved, noncellular vascular outlines and the underlying viable tumor ( Figure 2 , Middle left). The vessels within the viable tumor and abutting the necrotic area were massively hyalinized, with very narrow lumens.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


