Ranibizumab (RanibizumabTM) in the Treatment of Wet Macular Degeneration
Monica Rodriguez-Fontal
Virgil D. Alfaro III
John B. Kerrison
INTRODUCTION
Pathologic angiogenesis and vascular leakage have been recognized as major causes of visual loss in the wet form of age-related macular degeneration (AMD) (1). Although a combination of molecular regulators is involved in this complex process (2), multiple studies evidence the role of the vascular endothelial growth factor (VEGF) as the main signal in the ocular neovascularization (3,4 and 5).
The VEGF, also known as VEGF-A, is a heparin-binding glycoprotein that belongs to a gene family that includes VEGFC, VEGF-D, VEGF-B, and placenta growth factor (PIGF) (1). The human VEGF-A has 4 different isoforms as a result of the alternative exon splicing, VEGF121, VEGF165, VEGF189, VEGF206; the numbers show how many amino acids the VEGF molecule has after each cleavage. Other less frequent variants that have been reported are VEGF145 and VEGF183 (1). The VEGF165 is the major isoform (6) (Fig. 35-1). Inhibiting the action of the VEGF (the isoform165 or all the isoforms) has been postulated as a possible treatment in several diseases including solid tumors and ocular diseases that involved visual loss as a result of vascular leakage or pathologic angiogenesis.
The use of VEGF inhibition as a treatment for human disease has been studied for many years, and different therapeutic agents targeting the VEGF have been developed. A humanized monoclonal anti-VEGF antibody, Bevacizumab (Avastin®; Genentech, CA) was approved by the FDA in 2004 for intravenous therapy of metastatic colorectal cancer; in early 2004, Rosenfeld et al, initiated the use of bevacizumab in the treatment of wet AMD. In their first study SANA (Systemic Avastin for Neovascular AMD) the investigators used the drug intravenously and subsequent studies have used it, off-label, with intravitreal injections (7,8,9 and 10).
Macugen® (pegaptanib sodium; Eyetech Pharmaceuticals, NY), an anti-VEGF aptamer, was the first anti-VEGF agent proved to be efficacious in patients with wet AMD (11); its complex structure allows it to specifically bind the VEGF165 isoform.
In June 2006, the U. S. Food and Drug Administration (FDA) approved ranibizumab for the treatment of neovascular wet AMD. Ranibizumab (LucentisTM; Genentech, CA) is a recombinant, humanized, monoclonal anti-VEGF antibody fragment, with the ability to bind to all the isoforms of the VEGF (12) (Fig. 35-2).
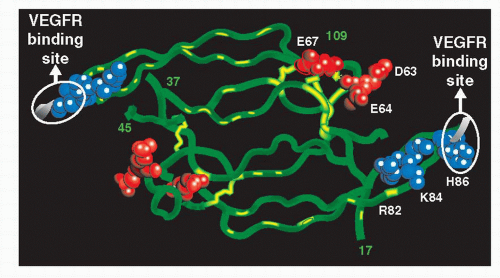 Figure 35-1. Vascular endothelial growth factor (VEGF), a homodimeric glycoprotein that is secreted in response to hypoxia and ischemia. VEGF induces angiogenesis and vascular permeability. Arrows show the binding site VEGF receptor. |
This chapter reviews the science and clinical trials of LucentisTM (ranibizumab) and the role of this drug in the treatment of subfoveal choroidal neovascularization (CNV) secondary to AMD.
BACKGROUND
The observation that solid tumor growth can be accompanied by increase vascularity was made one century ago (1). Ide et al. (13) postulated in 1939 the existence of a tumor-derived blood vessel growth-stimulating factor that may be responsible for inducing the neovascularization in the growing tumor. In 1948, Isaac Michaelson (14) proposed that a diffusible angiogenic “factor X” produced by the retina may be responsible for the retinal and iris neovascularization that occurs in proliferative diabetic retinopathy and other retinal disorders, such a central vein occlusion. In 1971, Folkman et al. (15) proposed that anti-angiogenesis might be an effective treatment for human cancer and initiated the efforts to isolate a “tumor angiogenesis factor.” In 1983, Senger et al. (16) described the partial purification of a tumor protein able to induce vascular leakage, named “tumor vascular permeability factor.” Because it was not isolated and sequenced, it remained molecularly unknown at that time. In 1989, Ferrera et al. (17) reported the isolation of a diffusible endothelial cell-specific mitogen, named VEGF; that was demonstrated later to be the same molecule that Senger found. After these initials steps, numerous papers have been published supporting the theory that VEFG is an endothelial cell mitogen and vascular permeability factor.
Aiello et al. (5) and Malecaze et al. (18) reported elevations of the VEGF in the aqueous humor and vitreous humor of the human eyes with proliferative retinopathy secondary to diabetes and other retinal disorders, demonstrating a temporal correlation between the VEGF elevations and the active proliferative retinopathy.
Studies by Kvanta et al. (19) and Lopez et al. (20) suggest a role for VEGF in the progression of the CNV in AMD; the authors
demonstrated the inmunohistochemical localization of the VEGF in surgically resected CNV membranes from AMD patients.
demonstrated the inmunohistochemical localization of the VEGF in surgically resected CNV membranes from AMD patients.
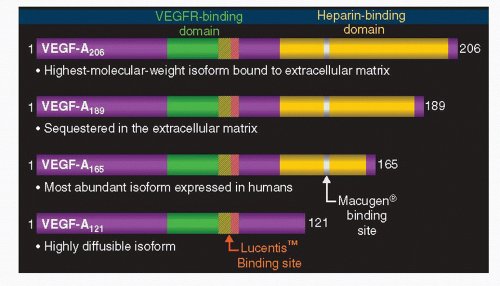 Figure 35-2. Ranibizumab (RanibizumabTM Genentech, CA) is a recombinant, humanized, monoclonal anti-vascular endothelial growth factor (VEGF) antibody fragment, with ability to bind to all the isoforms of the VEGF. (From Ferrara et al. Nat Med 2003;9:669; Robinson and Stringer. J Cell Sci 2001;114:853, with permission; Macugen® (pegaptanib) Pl 2005. Genetech, data on file.). |
Significant experimental evidence began to support the theory that VEGF inhibition could lead to suppression of ocular angiogenesis. Aiello et al. (21) evaluated in vitro and in vivo the inhibition of the VEGF and subsequent CNV using VEGF-neutralizing chimaeric proteins in a murine model of ischemic retinopathy. With this study Aiello clearly demonstrated that stimulation of the bovine retinal endothelial cell growth by both exogenous VEGF and hypoxia-induced VEGF was effectively suppressed with in vitro use of VEGF-receptor chimaeric proteins. Furthermore, he demonstrated, using a highly reproducible murine model of ischemia-induced retinal neovascularization, that inhibition of the VEGF reduces retinal neovascularization in vivo. Aiello (21) also highlighted another interesting theory, when he realized that the VEGF-neutralizing chimaeric proteins incompletely inhibited retinal neovascularization. The author theorized that penetration of the VEGF-neutralizing chimaeric proteins (human Flt-IgG and murine Flt-1-IgG both consist of entire VEGF receptors bound to IgG heavy chain) into the retinal tissue was unlikely, because of the size of the bulky chimaeric proteins, limiting the protein actions to the capillaries on the inner retinal surface.
Adamis et al. (4) also demonstrated that the VEGF was necessary for the retinal ischemia-associated iris neovascularization in a monkey model. The authors prevented iris neovascularization in monkey eyes with injections of an anti-VEGF monoclonal antibody.
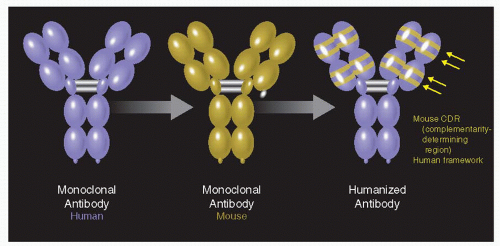 Figure 35-3. The humanization of muMAb VEGF A.4.6.1 involved the transfer of six complimentary-determining regions (CDR) from muMAb VEGF A.4.6.1 to a human framework by site-directed mutagenesis. The final antibody is a rhuMAb (recombinant, humanized, monoclonal antibody). |
Synthesis of Ranibizumab: Humanization, Affinity-Maturation, and Fragmentation
Murine antibodies (muMAb VEGF) are effective in the primate model (4), but their use as a human therapy was limited by the anti-globulin secondary immune reaction (22,23). Chimaeric molecules, where human constant regions are fused with variable rodent domains, are still capable of eliciting an immune response (24). Humanization of the murine monoclonal antibodies was an effective solution to overcome this clinical limitation. Presta et al. (25) described how the transfer of six complimentary-determining regions (CDR) from muMAB VEGF A 4.6.1 to a human framework by site direct mutagenesis and subsequently reduce its binding to VEGF over 1000 fold, humanizing the murine antibody (Fig. 35-3). To achieve binding equivalent to the original muMab, seven framework residues in the humanized variable light domain were changed from human to murine. Comparison of the humanized and the chimaeric antibodies revealed the same activity.
It remained to be investigated whether such recombinant proteins would reach their target following intravitreal administration. Mordenti et al. (26) suggested that rhuMAb VEGF Fab (fragment of the entire antibody with affinity for the
VEGF) might be a more effective therapy for AMD than fulllength rhuMAb antibody because of its ability to diffuse into the retinal-choroidal boundary. Mordenti et al. (26) studied the safety, the pharmacokinetics, and the retinal distribution of rhuMab HER2 (the full-length humanized antibody, 148 kD) and rhuMab VEGF Fab (the humanized Fab antibody 48.3 kD), following bilateral intravitreal injections in Rhesus monkeys. Retinal tissue distribution of 125I-labeled rhuMab HER2 and rhuMAb VEGF Fab was evaluated with microautoradiography. One hour post-injection, rhuMab VEGF Fab was detected throughout the retinal layers while rhuMab HER2 was found only in the vitreous cavity. RhuMab HER2 failed to penetrate the inner limiting membrane (ILM) (Fig. 35-4). RhuMab VEGF Fab was detected throughout the retinal layers as far as the retinal pigment epithelium (RPE) in the medial and lateral portions. By day 1, rhuMab VEGF Fab was evenly distributed throughout all of the retinal layers and remained so through day 4. Signals peaked to a maximum at day 1 and did not persist past day 7. Analysis of vitreous fluids revealed a halflife of 5.6 days of the full-length rhuMAb HER2 antibody and 3.2 days for the rhuMAb VEGF Fab, supporting the conclusion that the rate of vitreous clearing is directly affected by the molecular size of the molecules. Plasma levels of rhuMAb VEGF Fab remained constant below the assay limit of detection (7.8 ng/ml) throughout the study. Plasma levels of rhuMAb HER2 ranged from <4% to 30%. The authors suggested that Bruch’s membrane may act as a barrier for rhuMAb VEGF fab, preventing further diffusion into the choroid and resulting in an accumulation of fragmented antibody in the RPE. The ILM may also act as a physical barrier to the full-length antibody. Based on this assay, rhuMAb was fragmented to obtain Fab fragment with the same affinity as the whole antibody but with greater ability for penetration in the tissue (Fig. 35-5).
VEGF) might be a more effective therapy for AMD than fulllength rhuMAb antibody because of its ability to diffuse into the retinal-choroidal boundary. Mordenti et al. (26) studied the safety, the pharmacokinetics, and the retinal distribution of rhuMab HER2 (the full-length humanized antibody, 148 kD) and rhuMab VEGF Fab (the humanized Fab antibody 48.3 kD), following bilateral intravitreal injections in Rhesus monkeys. Retinal tissue distribution of 125I-labeled rhuMab HER2 and rhuMAb VEGF Fab was evaluated with microautoradiography. One hour post-injection, rhuMab VEGF Fab was detected throughout the retinal layers while rhuMab HER2 was found only in the vitreous cavity. RhuMab HER2 failed to penetrate the inner limiting membrane (ILM) (Fig. 35-4). RhuMab VEGF Fab was detected throughout the retinal layers as far as the retinal pigment epithelium (RPE) in the medial and lateral portions. By day 1, rhuMab VEGF Fab was evenly distributed throughout all of the retinal layers and remained so through day 4. Signals peaked to a maximum at day 1 and did not persist past day 7. Analysis of vitreous fluids revealed a halflife of 5.6 days of the full-length rhuMAb HER2 antibody and 3.2 days for the rhuMAb VEGF Fab, supporting the conclusion that the rate of vitreous clearing is directly affected by the molecular size of the molecules. Plasma levels of rhuMAb VEGF Fab remained constant below the assay limit of detection (7.8 ng/ml) throughout the study. Plasma levels of rhuMAb HER2 ranged from <4% to 30%. The authors suggested that Bruch’s membrane may act as a barrier for rhuMAb VEGF fab, preventing further diffusion into the choroid and resulting in an accumulation of fragmented antibody in the RPE. The ILM may also act as a physical barrier to the full-length antibody. Based on this assay, rhuMAb was fragmented to obtain Fab fragment with the same affinity as the whole antibody but with greater ability for penetration in the tissue (Fig. 35-5).
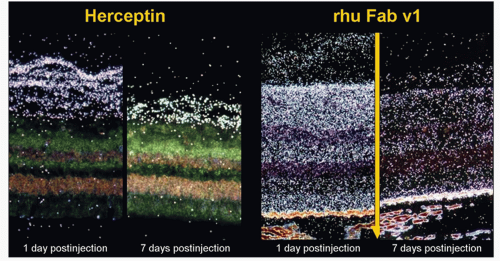 Figure 35-4. Histoautoradiograph of 125I-rhuMab VEGF Fab V1, the humanized Fab antibody (Column B) and 125Iodine (I)-rhuMab HER2, the full-length humanized antibody (Column A) following bilateral intravitreal injections in Rhesus monkeys. The Fab fragment penetrated all of the layers of the retina evenly extending as far back as the retinal pigment epithelium while the full-length antibody failed to penetrate farther than the ILM at any point during the study. |
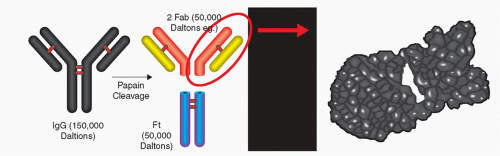 Figure 35-5. Papain cleavage of IgG antibody into two Fab fragments and the Fc fragment. The Fab portion is approximately a third of the original IgG. |
Following humanization, the Fab portion of the antibody (known as rhuMAb VEGF or recombinant humanized Monoclonal Antibody VEGF) was affinity-matured through CDR
mutation and affinity selected by monovalent phage display (27). It was possible to improved contacts between antibody and antigen. Two mutations resulted in increased van der Waals contact and improved hydrogen bonding (Figs. 35-6 and 35-7). The final antibody (affinity-matured rhuMAb) has improved affinity for several VEGF variants as compared with the parental antibody.
mutation and affinity selected by monovalent phage display (27). It was possible to improved contacts between antibody and antigen. Two mutations resulted in increased van der Waals contact and improved hydrogen bonding (Figs. 35-6 and 35-7). The final antibody (affinity-matured rhuMAb) has improved affinity for several VEGF variants as compared with the parental antibody.
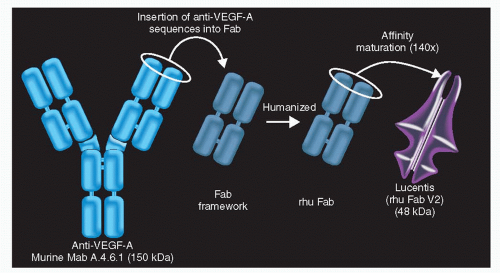 Figure 35-6. RhuMAb vascular endothelial growth factor (VEGF) was fragmented and affinity matured through complementarity-determining region (CDR) mutation and affinity selection by monovalent phage display. |
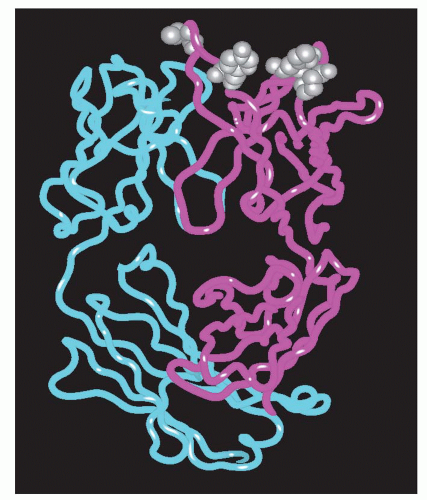 Figure 35-7. The vascular endothelial growth factor (VEGF) and the affinity-matured Fab fragment complex. Locally, it was possible to improve the contact between the antibody and the antigen through two mutations that improved the hydrogen bonding and van der Waals contact. |
PHARMACOKINETICS AND MECHANISM OF ACTION OF RANIBIZUMAB
Pharmacokinetics
Graudreault et al. (28) studied the ranibizumab (rhuFabV2, the rhuFab with affinity-matured) pharmacokinetic (PK) profile in cynomolgus monkeys. The selection of these monkeys was based on the idea of using the same animal model that was used to characterize the ranibizumab profile. The authors found that ranibizumab clears equivalently from all ocular compartments, with a half life of 3 days; 50% of the drug reached the circulation after 2 days, consistent with the half life and also suggesting that intraocular metabolism does not play a significant role in the elimination of ranibizumab from the vitreous. The authors observed the ability of the drug to penetrate the retina in cynomolgus monkeys, a finding consistent with the previous study in Rhesus monkeys using the first generation of rhuFab (rhuFabV1 without maturation) (26).
The same results were obtained after intravitreal administration of ranibizumab in rabbits. Ranibizumab clears from rabbits’ vitreous cavity in 2.9 days (29). The distribution of 125I-labeled ranibizumab determined by microautoradiography was consistent with specific sites of VEGF production and binding (ganglion cell and photoreceptor layer) (29). Avery et al. have shown that ranibizumab penetrates the full thickness retina at 1 to 3 days (30). In patients with neovascular AMD the pharmacokinetic profile was investigated following monthly intravitreal administration of ranibizumab; the maximum ranibizumab serum concentrations were low (0.3 ng/ml to 2.36 ng/ml). These levels were below the concentration of ranibizumab (11 ng/ml to 27 ng/ml) thought to be necessary to inhibit the biological activity of VEGF-A by 50%, as measured in an in vitro




Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
