Primitive Neuroectodermal Tumors
In this chapter, we describe neoplasias of several neural, neuroglial, and neuroganglionic tissues that share an ancestry from the primitive epithelium of the neural tube. This heterogeneous group may involve the orbit as primary, secondary, or metastatic growths.
Retinoblastoma
Retinoblastoma develops from primitive embryonal retinal cells, either photoreceptors or neuronal types. The tumor may display evidence of photoreceptor differentiation (Flexner-Wintersteiner rosettes) or show areas of glial differentiation (Atchaneeyasakul and Murphree, 2001; Biswas and Shanmugam, 1996). As in the previous editions of this text, our discussion chiefly concerns the extraocular and orbital extension of the intraocular retinoblastoma.
Incidence
Calculation of the true incidence of retinoblastoma is complex. There are many variables to consider, such as the size of the sample (a minimum of 100 cases), the age and sex of the children, their race, analysis of the data, basis of diagnosis (clinical or pathologic), unilateral or bilateral involvement, hereditable or spontaneous type, and estimation of the population at risk. Most commonly, the incidence is reported as the number of cases of tumor diagnosed in a period per live births for that period (Suckling et al., 1982). The lack of a worldwide uniform tumor registration protocol makes it difficult to attain an accurate calculation of incidence.
In the 40-year period between 1931 and 1971, many estimates of the frequency of retinoblastoma were published. In a population-based study, Bishop and Madson (1975) compiled a list of some of the publications during the 40-year time period (see Table 9.1).
We have not examined each publication in this table to determine which of the variables listed in the prior paragraph were or were not included in their survey. Retinoblastoma appears to have become more frequent between 1951 and 1971.
Tamboli et al. (1990) estimated the incidence of retinoblastoma in the United States from data on 220 cases in the files of the National Cancer Institute in Bethesda, Maryland (1974 through 1985) and compared these cases with previous US population studies. They concluded the incidence of retinoblastoma was almost uniform from 1974 to 1985. Riley (written communication, 2003) analyzed the 475 consecutive cases of retinoblastoma (1983 to 2002) in the registry of the King Khaled Eye Specialist Hospital, Riyadh, Saudi Arabia. This registry includes only children of Saudi Arabian descent. He concluded that, over the 19-year period, there had been no change in the incidence of retinoblastoma in their population-based study.
A register-based study of male and female patients born in the Netherlands between 1862 and 1995 was undertaken by Moll et al. (1997). Nine hundred and ninety-five patients were analyzed by a group of nine authors, including ophthalmologists, biostatisticians, epidemiologists, geneticists, and functional morphologists. Since 1945, the incidence of retinoblastoma appears to have stabilized (1 per 17,000 live births). Further, no significant differences in the incidence between males and females were found.
If the true incidence of retinoblastoma in the United States has not changed significantly in the past several decades, it is reassuring to believe that the retinoblastoma gene is not mutating to increase the number of cases of the tumor.
Table 9.1 Frequency of Retinoblastoma | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
In the 7-year interval between the first and second editions of Orbital Tumors (1973 and 1980), 1,100 cases of intraocular retinoblastoma were observed and treated (Rootman et al., 1978; and others). This collection of data corresponded to the era when enucleation was the preferred treatment of unilateral retinoblastoma. Orbital involvement of retinoblastoma varied from 8% to 10% of the total sample.
Soon thereafter, chemotherapy superseded enucleation as the primary treatment for intraocular retinoblastoma, and the number of enucleated eyes available for pathologic studies diminished. In the past 2 decades, many eyes have been salvaged that otherwise would have been enucleated. However, not all eyes with orbital involvement respond to conservative, nonsurgical therapy, and the affected eye must be removed. Besides, such eyes have been enucleated at other institutions. Earlier diagnosis of intraocular tumors has likely reduced the number showing orbital involvement. Neither sex has a higher incidence of retinoblastoma; unilateral retinoblastoma occurs more than three times as often as bilateral disease (Pendergrass and Davis, 1980), and incidence of bilateral disease is the same in whites and blacks (Tamboli et al., 1990).
Most retinoblastomas are diagnosed before the patient reaches the age of 3 (Robertson, 2003) to 4 (Zimmerman, 1985) years. However, Shields et al. (1991) noted that 8.5% of patients among their 400 consecutive cases with retinoblastoma were older than 5 years.
Clinical Features
In the United States, leukocoria, often associated with strabismus, is the principal presenting sign of retinoblastoma. Orbital extension occurs sometime after or during treatment of the intraocular tumor. The presenting symptom is a painless proptosis that heralds a retrobulbar mass. If an implant has been placed in the orbital socket after enucleation, the need for frequent adjustment of the prosthesis or its extrusion is indicative of an orbital mass. A vitreous hemorrhage in an older child may conceal an unsuspected retinoblastoma that is discovered after a vitrectomy for a nonclearing hemorrhage. In developing countries, retinoblastoma may present as an orbital tumor rather than an intraocular lesion. In such cases, a large, reddish black, friable mass may bulge out beyond the confines of the eyelid.
Imaging Aspects
Although >90% of intraocular retinoblastomas show intralesional calcification with computed tomography (CT) scan, calcification is rarely present in the extraocular extension (Kaufman et al., 1998). Nevertheless, CT scan shows the size and position of the retrobulbar mass. Magnetic resonance imaging (MRI) may be more useful in determining the degree of optic nerve involvement. The orbital extension is hyperintense on T1-weighted images-weighted images and hypointense on T2-weighted images-weighted images.
Pathology
Figures 9.1 and 9.2 illustrate two of the routes of extraocular extension of an intraocular retinoblastoma. Other routes of escape are the short and long ciliary nerves and the direct breaching of the wall of the eye manifested by episcleral nodules. When such bluish pods are visible, it is likely that subclinical microscopic spread of retinoblastoma has occurred into the retrobulbar tissue. Invasion of the choroid by the intraocular mass is probably a source for distant metastasis that may precede direct orbital extension. The presence of tumor along the line of transection of the optic nerve or tumor cells along the pial surface of the nerve suggests probable intracranial seeding of neoplasm.
The retinoblastoma is neuroblastic in origin and may arise in any of the nucleated layers of the retina. Immunohistochemical studies with interphotoreceptor retinoid-binding protein antibodies and with neuron-specific enolase support the neuronal origin of the tumor (Rodrigues et al., 1987). Glial differentiation, although demonstrable in tissue cultures of retinoblastoma cells (Kyritsis et al., 1984),
has not been convincingly demonstrated in intraocular tumor cells (Lane and Klintworth, 1983) or within orbital extensions of the tumor.
has not been convincingly demonstrated in intraocular tumor cells (Lane and Klintworth, 1983) or within orbital extensions of the tumor.
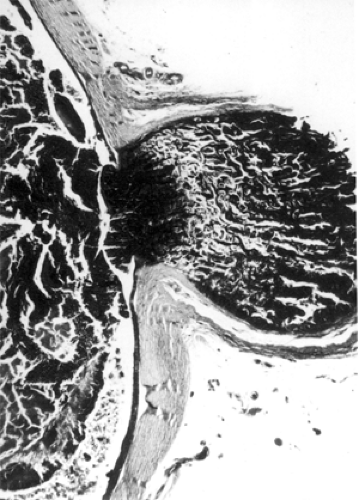 Figure 9.1 Orbital involvement most commonly results from penetration through the lamina cribrosa and spread in the optic nerve (original magnification × 10). |
The intraocular tumor may show varying degrees of differentiation and the formation of fleurettes and rosettes of the Flexner-Wintersteiner type, representing an attempt to produce photoreceptor cells. This high degree of differentiation is not observed in the orbital extension of the tumor, and the small dark cells with their hyperchromatic nuclei and multiple mitotic figures are dispersed in sheets (see Fig. 9.3). Necrosis, with or without calcification, is a prominent feature, and DNA liberated from the necrotic tumor cells is seen as a blue ring in the blood vessel walls with the hematoxylin and eosin stain.
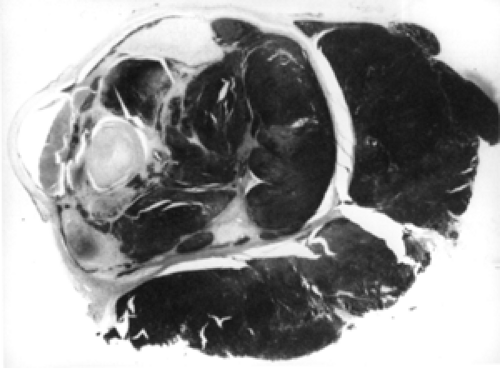 Figure 9.2 Massive orbital invasion apparent along the emissary vascular channels rather than through the optic nerve (original magnification × 2½. |
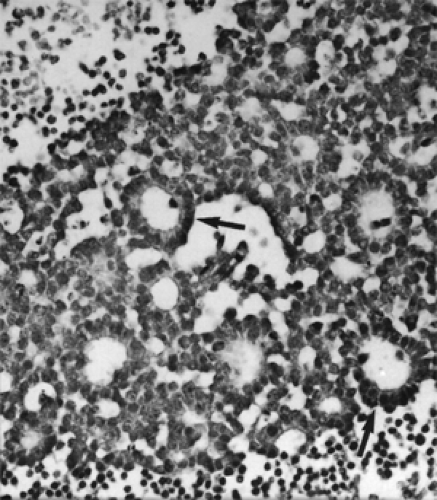 Figure 9.3 A sheet of small, dark cells with hyperchromatic nuclei and Flexner-Wintersteiner rosettes (arrows) bordered by areas of necrosis (upper left and lower right) (original magnification × 400). |
Management
Commencing in the late 1980s and early 1990s, chemotherapy became the preferred initial therapy for the nonsurgical management of retinoblastoma. Our third edition of Orbital Tumors (1994) noted the drug combination protocols in use for cases of intraocular, orbital, and intracranial tumors. Gradually, adjustments such as local therapy applied to the intraocular mass and radiotherapy to the overall orbit have been added to treatment protocols.
Local (“focal”) treatment to the intraocular tumor may include either hyperthermia or photocoagulation through the transpupillary route and/or cryotherapy and brachytherapy applied to the exterior of the globe. Finally, external beam radiotherapy to the globe and orbit may be necessary. Focal therapy alone may be given if the diagnosis is early and the intraocular tumor is small enough to be within the tolerance level of the eye to the local agent chosen for administration.
However, our main concern in this text is the treatment of patients with invasion of the orbit, the optic nerve, or both. This invasion occurs at the most advanced stage of local disease. The rationale for the initial use of chemotherapy is the observation that it sensitizes the
tumor bulk to the adjuvant modalities administered during or after chemotherapy. This mode is termed chemoreduction. At present, there is no uniform standard of the type or number of chemical agents used or the duration of the cycles of chemotherapy administered in a given time span. Some physicians recommend a combination of these drugs, that is, carboplatin, vincristine, and etoposide; others combine just two of these agents. Some physicians defer the use of a focal adjuvant on the intraocular tumor until some reduction in the overall bulk of the neoplasm by chemoreduction is observed. Others combine focal and systemic chemotherapy simultaneously. Whichever type of protocol is elected, it should be administered in close interaction with a pediatric oncologist who is knowledgeable about the adverse effects of these powerful chemical agents.
tumor bulk to the adjuvant modalities administered during or after chemotherapy. This mode is termed chemoreduction. At present, there is no uniform standard of the type or number of chemical agents used or the duration of the cycles of chemotherapy administered in a given time span. Some physicians recommend a combination of these drugs, that is, carboplatin, vincristine, and etoposide; others combine just two of these agents. Some physicians defer the use of a focal adjuvant on the intraocular tumor until some reduction in the overall bulk of the neoplasm by chemoreduction is observed. Others combine focal and systemic chemotherapy simultaneously. Whichever type of protocol is elected, it should be administered in close interaction with a pediatric oncologist who is knowledgeable about the adverse effects of these powerful chemical agents.
Sooner or later, these patients have external beam radiotherapy. At Mayo Clinic, the radiation equivalent of 45 Gy has been administered over a period of years. Irradiation should be used within 6 months of chemotherapy. If brachytherapy has been used during the course of chemotherapy, the total dose of irradiation should be reduced. Schvartzman et al. (1996) believe adjuvant irradiation is of value in preventing metastasis in patients who had invasion of the optic nerve. If these complex treatment protocols fail or disease recurs after an initial favorable response, enucleation is the last resort. Enucleation is also indicated for patients who, on presentation, have advanced retinoblastoma, that is, more than half the retina is affected by tumor, subretinal fluid and vitreous seeding are present, and visual acuity is markedly reduced. From reports in East Indian journals, eyes with advanced retinoblastoma at the time of presentation are almost the norm.
Prognosis
We have been pleased to read many publications in the literature from the 1990s and the early years of this century that emphasize the increasing survival of patients with retinoblastoma. This increased survival parallels advances and refinements in chemotherapy and its adjuvants over this time span. Robertson (2003) estimates that, at present, 95% of patients survive the malignant neoplasm. This estimate is probably based on patients with an intraocular tumor that has not spread beyond the interior of the eye.
Estimating the survival of patients with orbital extension (including the optic nerve) is unreliable. First, the number of cases of advanced retinoblastoma in North America is relatively small compared with the numbers in India and Africa. Second, these few cases have been treated with a variety of protocols, that is, from 2 to 12 cycles of chemotherapy with or without focal adjuvants. Questions remain about whether adjuvant therapy was administered simultaneously with or after chemotherapy. The same variables can be applied to the use of radiotherapy. Finally, some cases have been treated only with external beam radiotherapy (no chemotherapy). Trying to analyze such a hodgepodge is akin to comparing the proverbial apples and oranges. Robertson (2003) has noted that with extraocular extension and orbital involvement, a morbidity rate as high as 90% has occurred at 2-year follow-up.
What is needed now is an international study group composed of retinoblastoma experts who can establish standard protocols for the management of orbital diseases, provide a uniform system for analyzing data, and the required duration of follow-up observation.
Whether the ophthalmologist is dealing with local intraocular retinoblastoma or advanced disease, patients need to be kept under observation for a long time. Goto et al. (2002) reported recurrences of neoplasm 12 years after brachytherapy, and Shields et al. (2002) noted recurrence 25 years after external beam radiotherapy. Our fervent hope is that a new method of treatment will evolve that will eliminate the use of external beam radiotherapy in children with retinoblastoma.
Medulloepithelioma
Synonyms for medulloepithelioma are “diktyoma,” “teratoneuroma,” and “teratoid malignant medulloepithelioma.” This neoplasm may arise from the primitive neuroepithelium of the forebrain, along the course of the optic nerve, or from the intraocular ciliary body. The neoplasm has a multipotential cellular diversity, may be benign or malignant, and nonteratoid or teratoid type. A teratoid type contains one or more heteroplastic elements such as brain tissue and cartilage. Most publications discuss the medulloepithelioma in its most common location, the ciliary body. Here, most neoplasms are benign. A minority is malignant, but the degree is less than the tumors of the brain and optic nerve. Our concern, in this chapter, is the medulloepithelioma arising in the optic nerve. From this source, the tumor may extend into the orbital space or the intracranial vault, or retrograde into the eyeball.
Incidence
A medulloepithelioma of the optic nerve is very rare. This neoplasm has not been seen at Mayo Clinic during our 50-year collection of 1,795 consecutive, pathologically proved orbital tumors. Only a few well-authenticated cases originating in the optic nerve have been reported in the literature. Among these were a 4½-year-old boy (Reese, 1957), a newborn (Andersen, 1971), a 6-year-old girl (Green et al., 1974), a 2-year-old boy (O’Keefe et al., 1997), and a 3-year-old boy (Biswas et al., 1999). Harry and Morgan (1979) noted the neoplasm associated with a buphthalmic eye. Congenital cases have also been reported (Steinkuller and Font, 1997; Chidambaram et al., 2000).
Clinical Features
All reported cases have been unilateral and sporadic. All the children with medulloepithelioma, except those with congenital tumors, have almost the same presenting signs and symptoms as those with optic nerve glioma. Indeed, these cases were initially thought to be optic glioma until an invasive biopsy was performed. The presenting signs and symptoms include proptosis, displacement of the eye, some impairment of ocular motility, a dilated pupil, and some ophthalmoscopic abnormality of the optic disk. The intraocular tension may be elevated in some cases. However, at the time of discovery, children with medulloepithelioma tend to be a bit younger than the average age of children with optic glioma.
Imaging Aspects
In the article by Biswas et al. (1999), the CT scan of a 3-year-old patient (see Fig. 9.4) shows a well-defined, intraconal, homogeneous, hyperdense, enhancing lesion involving the optic nerve with bowing of the medial orbital wall that extends intracranially through the apex of the orbit and involves the left cavernous sinus. Axial T1-weighted MRI showed the mass to be hypodense.
Pathology
These tumors contain cellular elements that closely resemble medullary epithelium and are derived from the optic vesicle, optic cup, retinal pigment epithelium, nonpigmented and pigmented ciliary epithelium, vitreous, and neuroglia. The color of the neoplasm may depend on the content of pigmented ciliary epithelium (Spencer and Rao, 1996). All tumor cells have variable cytologic characteristics.
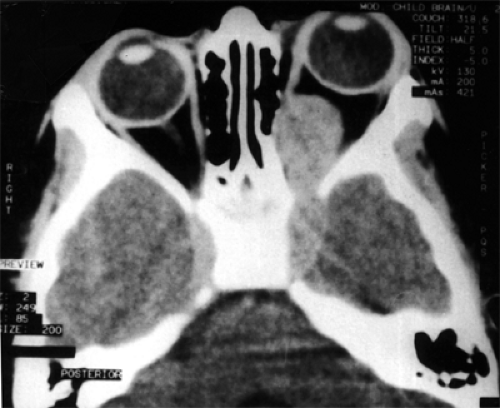 Figure 9.4 Computed tomography scan showing a well-defined, intraconal, homogeneous, hyperdense, enhancing mass extending intracranially through a widened orbital fissure and involving the left cavernous sinus. (From Biswas J, Bhushan B, Jayakumar N, et al. Teratoid malignant medulloepithelioma of the optic nerve: Report of a case and review of the literature. Orbit. 1999;18:191–196 , with permission.) |
The heterogeneous cellular makeup of any given medulloepithelioma may depend on its location. Tumors of the optic nerve are prone to contain more undifferentiated elements such as those seen in retinoblastoma. Malignant tumors are more common than benign lesions in this location. The components of a neoplasm affecting the ciliary body are composed of more mature cellular components. Here benign medulloepitheliomas are more common than malignant types.
The proliferating medullary epithelium is arranged in cords and sheets separated by cystic spaces and lined by a single layer of epithelium. These cysts contain hyaluronic acid (see Fig. 9.5). If these cysts are located on the surface of the tumor, they may break off and become free-floating in the vitreous. Malignant tumors may not differ significantly from benign lesions. Invasiveness and number of mitotic figures are criteria for malignancy.
Management
There is no standard pattern of management for an orbital medulloepithelioma. Too few cases are available to judge the effectiveness of exenteration, chemotherapy, and radiotherapy, either alone or in combination. The 6-year-old patient reported by Green et al. (1974) was living without metastasis 18 months after exenteration of the orbit. The 4-year-old patient described by Reese (1957) underwent enucleation. Nine months later, there was an orbital recurrence. A second surgery was performed
and irradiation was given. No evidence of tumor was found 8 months later. The 2-year-old boy reported by O’Keefe et al. (1997) underwent enucleation because of a presumed retinoblastoma. On histologic study, a medulloepithelioma of the optic nerve was found. Further tumor resection was performed. Radiotherapy and chemotherapy were also administered. Four years later, there was no evidence of recurrence. The 3-year-old patient reported by Biswas et al. (1999) was treated with external beam radiotherapy of 4,600 Gy in 20 fractions. No surgery was performed. Seventeen months after surgery the child was well without systemic metastasis but a month later brain metastasis occurred and the child died.
and irradiation was given. No evidence of tumor was found 8 months later. The 2-year-old boy reported by O’Keefe et al. (1997) underwent enucleation because of a presumed retinoblastoma. On histologic study, a medulloepithelioma of the optic nerve was found. Further tumor resection was performed. Radiotherapy and chemotherapy were also administered. Four years later, there was no evidence of recurrence. The 3-year-old patient reported by Biswas et al. (1999) was treated with external beam radiotherapy of 4,600 Gy in 20 fractions. No surgery was performed. Seventeen months after surgery the child was well without systemic metastasis but a month later brain metastasis occurred and the child died.
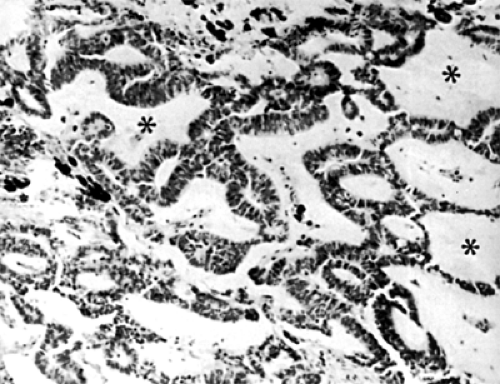 Figure 9.5 Typical cords of medullary epithelium separated by a zone of vitreous-like tissue (asterisks) in the optic nerve of a 5-year-old girl (hematoxylin and eosin, original magnification × 380). (From Spencer WH, Rao N. Medulloepithelioma. In: Spencer WH, Ophthalmic pathology: An atlas and textbook, Vol. 1, 4th ed. Philadelphia, PA: WB Saunders; 1996:607–608 , with permission.) |
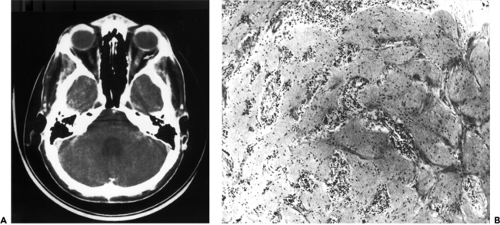 Figure 9.6 Metastatic medulloblastoma. A: Axial computed tomography scan demonstrates enlargement of the optic nerve (arrow). B: Subsequent biopsy revealed a malignant small cell neoplasm infiltrating the optic nerve that was consistent with metastasis (original magnification × 100). |
Medulloblastoma
Medulloblastoma is a neoplasm of the cerebellum and is not to be confused with medulloepithelioma. The medulloblastoma arises from the fetal granular layer of cells that covers the cerebellar cortex and is present at birth. Normally, this layer disappears during the first year of life. Metastasis from a medulloblastoma to the intraorbital portion of the optic nerve accounts for the one case of this tumor type in our orbital tumor study. This was a 19-year-old man who underwent removal of a midline medulloblastoma of the cerebellum in January 1984 (Garrity et al., 1989). Postoperatively, the patient was treated with radiotherapy, 55 Gy to the posterior cranial fossa and 37 Gy to the spinal cord. In April 1986, decreasing vision of the left eye was noted. Two months later, a visual acuity of 20/400 in the left eye was recorded. There was marked restriction of the visual field in the left eye, and 4 diopters of papilledema were noted. A CT scan (see Fig. 9.6A) showed enlargement of the left optic nerve. A biopsy of the enlarged nerve revealed a malignant small cell neoplasm consistent with metastatic medulloblastoma (Fig. 9.6B). Another 50 Gy of radiotherapy was given to the left optic nerve, supplemented with chemotherapy. Extensive metastasis to the spinal cord was demonstrated in February 1988, and the patient died 5 months later.
Intraorbital Optic Nerve Glioma
The nerve of the anterior visual pathway extending from the optic papilla to the geniculate bodies is morphologically and functionally a tract of the central nervous system comparable to the white matter of the brain. This nerve tract is surrounded by the same coverings as the brain, and the extrinsic supporting structures are vascular connective tissue septa and neuroglial cells.
It is the abnormal proliferation of supporting neuroglial cells to which the term glioma has been applied. Gliomas may arise anywhere in the central nervous system. Our discussion of the subject mainly concerns the glioma of the optic nerve rather than the manifestations of glioma of the optic chiasm or optic tract. Furthermore, our emphasis is directed chiefly to the intraorbital portion of the optic nerve because it is the glioma in this location that most closely mimics the clinical manifestations of a true orbital tumor.
The present trend is to regard these gliomas as true neoplasms rather than as hamartomas.
Incidence
Our 40-year survey (1948 to 1987) of consecutive cases of orbital tumors, published in the third edition of Orbital Tumors (1994), encompassed the era in which enucleation of the eye was the preferred management of an intraorbital optic nerve glioma. Therefore, the neoplasm was available for histologic verification. There were 34 gliomas in this survey. At the time, this represented 2.4% of our total 1,376 tumors. The tumor was the second most frequent orbital tumor in children at that time. Sixty-eight percent of our gliomas were seen in the patients’ first decade of life. The age range of our patients was 2 months to 46 years, with a mean of 8.5 years and a median of 5.5 years.
Another massive review of this feature of the neoplasm was also published by Dutton (1994). His survey was based on 2,297 cases from the world’s literature up to 1992. The optic nerve glioma occurred in 1.5% to 3.5% of all orbital tumors. The sex distribution was equal. Overall, 70% of patients presented in the first decade of life. Twenty-nine percent of gliomas were associated with neurofibromatosis, and among neurofibromatosis patients, optic nerve gliomas were detected in approximately 15%. Since these publications, update reports on the frequency of orbital optic nerve glioma have declined, particularly in the Americas and Europe, because chemotherapy and radiotherapy have replaced enucleation of the eye as the preferred initial management option. However, in Asia and Africa, cases of advanced glioma at the time of presentation (painful, marked proptosis, and blindness) seem more common. Here, enucleation is the only possible management option.
Clinical Features
The presenting pattern of an intraorbital glioma is almost always one or a combination of a painless proptosis of one eye (see Fig. 9.7), some displacement of the eye, some degree of visual dysfunction, and a disturbance of ocular motility. If the child is too young for visual acuity assessment, a visual-evoked potential can indicate the degree of visual impairment. This test also serves as a baseline for judging the progress of the tumor in this age-group of children. The latency of the visual-evoked potential increases as the neoplasm enlarges (Ng and North, 2001). Other subsequent signs and symptoms of glioma are an afferent pupillary defect and pallor or papilledema of the optic disk, and strabismus may develop. In our small cohort of patients, papilledema was more common than pallor in the intraorbital glioma, but the reverse was noted in the chiasmal-type gliomas. Older children have a central scotoma and some degree of achromatopsia.
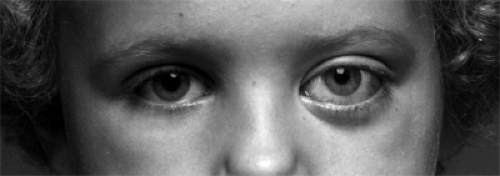 Figure 9.7 Upward displacement, subtle baring of the sclera, and slight proptosis of the left eye with vision reduced to light perception of 1 month’s duration in a 4-year-old girl. Chronic papilledema and moderate pallor of the left optic disk were also present. |
Imaging Aspects
At present, CT scan is the most useful imaging mode for the initial diagnosis of a presumed orbital optic nerve glioma. It pictures the size, shape, and extent of the neoplasm. Gliomas are usually globular (see Fig. 9.8), fusiform (see Fig. 9.9), or lobular in shape. The lobular glioma is the result of bending or buckling of the expanding nerve within the confined orbital space (see Fig. 9.10). The glioma is usually isodense, with minimal degrees of contrast enhancement, varying between 20% and 25%. The glioma is also well marginated because the dural covering, although stretched, contains the tumor within the nerve sheath. However, in long-standing gliomas, the density becomes less uniform and associated with areas of low attenuation suggesting a cyst-like degeneration (see Fig. 9.11).
MRI is best reserved for the follow-up period. It reveals changes in the size of the tumor and any extension of the mass toward the chiasm. The gliomas have slightly prolonged T1– and T2-weighted relaxation times
(Shen et al., 2001). As a result, the tumor on the T1-weighted image appears isodense or slightly hypointense compared with white matter. Areas of mucinous degeneration and necrosis appear hypointense. On T2-weighted images, the signal intensity may show greater variability but usually appears hyperintense compared with white matter.
(Shen et al., 2001). As a result, the tumor on the T1-weighted image appears isodense or slightly hypointense compared with white matter. Areas of mucinous degeneration and necrosis appear hypointense. On T2-weighted images, the signal intensity may show greater variability but usually appears hyperintense compared with white matter.
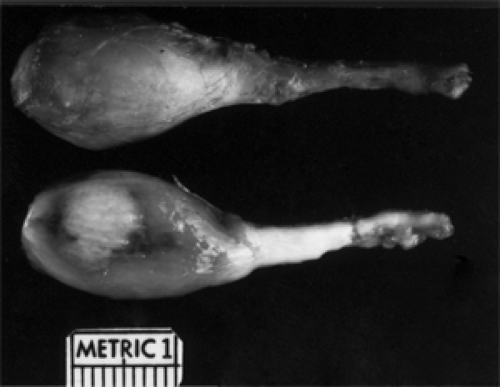 Figure 9.8 A globular glioma from a 12-year-old girl showing the external surface above and cut surface below. The rounded portion of the tumor (left) abutted and indented the back surface of the eyeball. |
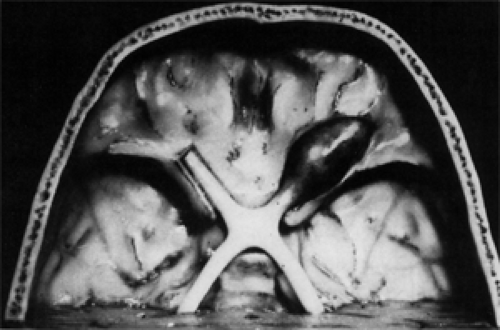 Figure 9.9 Model of a fusiform glioma of the orbital portion of the optic nerve. |
In lieu of a diagnostic incisional biopsy of the orbital glioma, several ophthalmologists and radiologists discuss their reliance on the differing radiographic features of optic nerve glioma and the orbital nerve sheath meningioma, particularly in older children and adults, for accurate diagnosis. The principal radiographic features of a meningioma surrounding the orbital optic nerve are a greater degree of contrast enhancement than a glioma, calcifications corresponding to the inherent psammoma bodies, and, of most importance, a characteristic tumor blush when studied by angiography, a feature absent in glioma. Unfortunately, some gliomas have an arachnoid hyperplasia that may give a false appearance of vascularization, thereby nullifying this assumed diagnostic clue (Liauw et al., 1996). A diagnosis of an orbital optic nerve neoplasm by radiographic imaging, even among those skilled in the nuances of imaging, is at best only presumptuous.
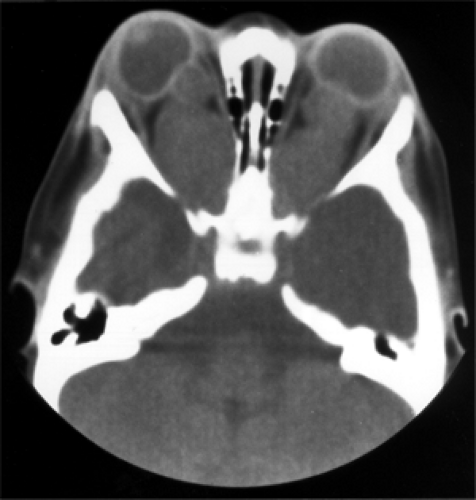 Figure 9.10 Axial computed tomography scan without contrast shows large, bilateral, isodense lobulated tumors filling the retrobulbar spaces and extending from the posterior margins of the eyeballs to the optic foramina. Note the indentation of the posterior wall of each eye by the tumors. The patient was a 5-month-old boy with bilateral ocular nystagmoid movement since birth. |
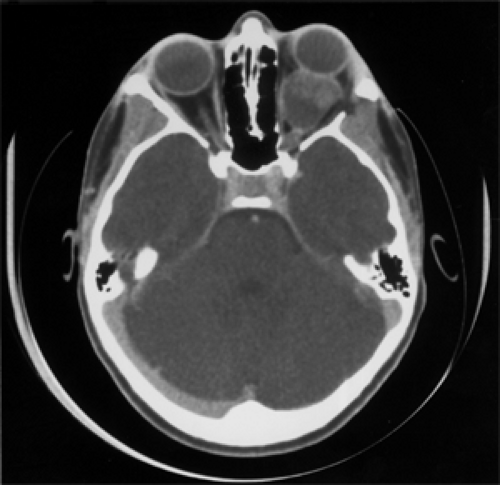 Figure 9.11 Axial computed tomography scan shows a large, well-marginated mass in the retrobulbar space of the left orbit associated with proptosis in a 3-year-old girl. The mass has areas of low attenuation adjoining areas of contrast enhancement suggesting cyst-like degeneration. There is no optic canal enlargement or chiasmal involvement. A biopsy diagnosis of optic nerve glioma was made when the patient was 6 years old. She received no subsequent therapy. At orbitotomy, the mass was a partially necrotic, gelatinous glioma with a large cyst-like component (coalescence of microcysts). |
Association with Neurofibromatosis
Over time, a number of publications have discussed this subject. We briefly summarize those reports that included an analysis of >100 cases.
In the article by Crowe and Schull (1953), 635 (29%) of 2,186 published cases with clinical data demonstrated signs of neurofibromatosis type 1 (NF1). The authors believed the true incidence of NF1 may be considerably higher because most studies failed to mention the presence or absence of this disease in their patients.
Chutorian et al. (1976) reported that patients with NF1 may have a higher incidence of bilateral optic gliomas than those without NF1.
Of the 318 patients described with glioma confined to the optic nerve by Imes and Hoyt (1986) and Listernick et al. (1989), only seven patients (2.2%) had bilateral disease with no detectable tumor crossing the chiasm.
Alvord and Lofton (1988) analyzed 155 cases of intraorbital optic nerve gliomas, including patients with and without NF1. None of the patients of either type who underwent a complete excision of the tumor with a microscopically clean proximal stump had a recurrence. However, when the surgical stump was not free of tumor cells or had not been examined at all, the tumor was prone to recurrence because of chiasmal spread. In patients without NF1, the recurrence rate was 5%. In those with NF1, the recurrence rate was 10%.
Pathology
The most common type of glioma affecting the orbital nerve in children is the juvenile fibrocystic astrocytoma, grade 1. It is benign and very slow growing. Its growth often comes to a spontaneous halt, and it remains an intraorbital mass. If growth continues, the neoplasm extends through the bony optic foramen and invades the intracranial portion of the optic nerve. The origin of the tumor is the glial cell of the intraneural, septal supportive network. The proliferating tumor cells may have vesicular or hyperchromatic nuclei (see Fig. 9.12). A minimal amount of cytoplasm may be evident in some cells.
Intracellular, irregularly shaped, eosinophilic processes may be present in association with thickened cytoplasmic glial filaments. These degenerative masses are Rosenthal fibers (see Fig. 9.13). Their presence is characteristic of a low-grade astrocytoma, but they are not specific for this tumor.
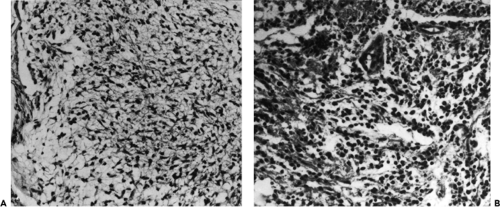 Figure 9.12 A: The glioma is composed of fine fibrillary astrocytes (original magnification × 155). B: Small, regular, rounded nuclei suggest the pattern of an oligodendroglioma (original magnification × 155). |
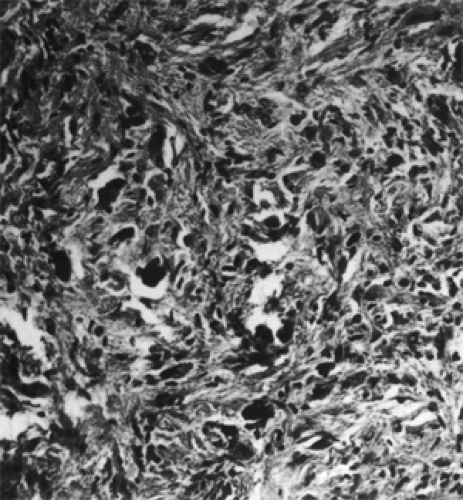 Figure 9.13 Grade 3 astrocytoma showing mitotic figures, increased cellularity, cellular anaplasia, and pleomorphism. Irregular-shaped, amorphous, dense-staining structures are Rosenthal fibers (original magnification × 155). |
Areas of the tumor remote from the vascular supply may undergo microcystic degeneration (see Figs. 9.13 and 9.14), and the foci of pooled mucopolysaccharide are visible on
CT scan. Microcalcospherites are occasionally seen within the stroma.
CT scan. Microcalcospherites are occasionally seen within the stroma.
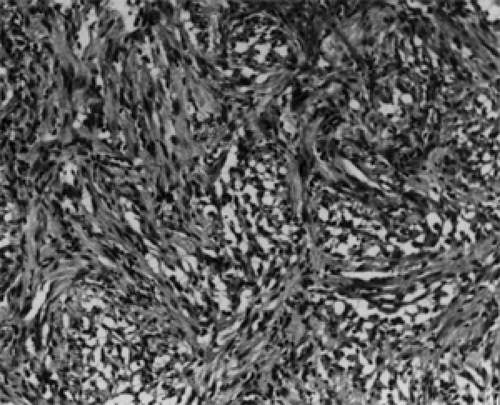 Figure 9.14 Grade 2 neoplasm is more cellular and cytoplasmic processes are coarser, imparting a more fibrous appearance. Numerous microcysts are evident (original magnification × 100). |
The tumor cells promote a reactive response of the leptomeningeal cells so that the pial septa thicken, as do the surrounding leptomeninges. This adds to the increasing diameter of the nerve (see Fig. 9.15). Several authors believe this hyperplasia is a feature indicative of associated NF and that intraneural growth without hyperplasia is characteristic of an isolated optic nerve glioma. In the past, biopsy material from the peripheral portion of the nerve has been misinterpreted as fibrous meningioma.
A less common type of glioma of the optic nerve occurs in adults (>20 years old). It is a highly malignant neoplasm, an anaplastic astrocytoma that is infiltrative, grows rapidly (early blindness), and causes death by growing proximally into the intracranial portion of optic pathways. The tumor shows nuclear pleomorphism, mitotic activity, endothelial proliferation, and necrosis. Its most common location is the chiasmal portion of the visual pathway.
Management and Course
If, on initial examination, a child has several signs or symptoms of an optic nerve glioma and a routine orbital CT scan shows a well-marginated mass within the confines of the intraorbital optic nerve, a presumptive diagnosis of a low-grade optic nerve glioma is made. If, on further analysis of the CT scan, there is no evidence of proximal expansion of the mass into the optic foramen, an incisional biopsy is not done.
The parents should be told that it is likely the tumor is benign and will not metastasize, but over succeeding months or years, complete loss of vision will occur. The tumor will grow slowly, and there will be no pain. The parents can also be told that the tumor, at this stage, could be surgically removed, but this would result in immediate and total blindness. Instead, we favor no treatment, thereby allowing preservation of vision as long as possible. We do not recommend radiotherapy or chemotherapy. We also prefer a follow-up visit in 6 months. At this time, an MRI scan can be performed to provide more precise information as to the presence or absence of intracranial extension of tumor. If negative, the child can be followed up at yearly intervals with successive MRIs. If at any time in the follow-up interval, invasion or extended growth along the nerve is shown, surgical removal is our choice of therapy. A frontal orbitocraniotomy with unroofing of the orbit can be performed. The superior wall of the long optic canal and a long piece (short of the chiasm) of the optic nerve and the tumor can be removed. With careful dissection, the eye can be salvaged. If the proximal end of the nerve is free of tumor cells microscopically, the patient can probably be cured.
Rush et al. (1982), studied 33 histologically verified optic nerve gliomas that had been removed between 1919 and 1973. Twenty-three patients underwent total excision of tumor without supplementary radiotherapy. Nine patients, whose tumors were incompletely excised, received supplementary radiotherapy. One patient received no therapy. Five of these patients died: Four of the nine who underwent incomplete surgical removal of tumors and the one patient with no treatment. The remaining 28 patients were still living at the conclusion of their follow-up. Their mean survival interval was 17 years; the longest survival was 44 years and the median was 16 years. Rush et al. (1982) rightfully concluded that survival was significantly associated with completeness of surgical excision.
The malignant optic nerve glioma is, chiefly, primary in the chiasm. Its incidence, clinical features, therapy, and course are beyond the scope of this review.
Over the many years that an intraorbital optic nerve glioma has been known, instances of its spontaneous regression have been occasionally noted. Parsa et al. (2001) reviewed 13 cases of spontaneous regression of optic gliomas documented by serial neuroimaging and collected internationally from 12 medical centers. All tumors met radiologic criteria for diagnosis. Three of the 13 cases showed involvement of the orbital portion of the optic nerve, either as an isolated tumor or a participant in thickening of the nerve from the eyeball to the chiasm. However, only one of these cases was verified histologically. The biopsy report was a pilocytic astrocytoma grade 1 in a 4-year-old girl with thickening of the entire right optic nerve. No treatment was offered. At the age of 17 years, her vision was 20/30 in the affected eye, and an MRI showed only trace enlargement compared with the left optic nerve. The other 10 cases in their series involved the optic chiasm. Some cases in the literature reporting regression of tumor after incomplete excision may have been surgically induced.
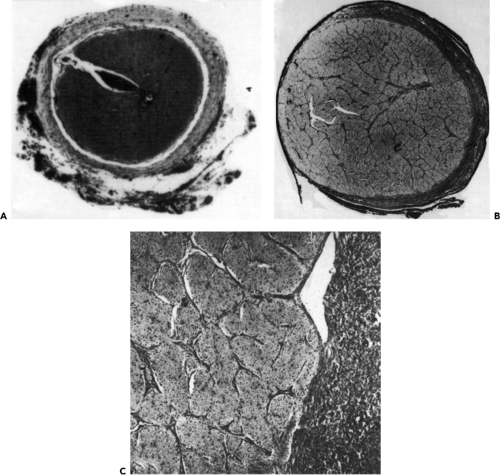 Figure 9.15 A: Normal optic nerve (original magnification × 6). B: Symmetric enlargement of the nerve reflected in the increased thickness of the individual nerve fascicles compared with (A) at the same magnification (original magnification × 6). C: Optic nerve (left) and perineural proliferation of the arachnoid cells and connective tissue (right) that is not part of an optic nerve glioma (original magnification × 4). |
Neuroblastoma and Ganglioneuroblastoma
These tumors are derived from the sympathogonia of neural crest origin that are destined to populate the sympathetic nervous system. If it can be said that the neuroblastoma represents a first step in maturation from the primitive neural crest cell, the ganglioneuroblastoma represents the second rung of the ladder of differentiation. Other primary sources are the thoracic, cervical, or pelvic sympathetic nervous system chain. Both tumors are considered malignant, but the mature third sibling in this family, the ganglioneuroma, is benign. Our further remarks chiefly concern the metastatic orbital neuroblastoma.
Incidence
In approximately 55% of cases, the primary site of the tumor is the adrenal gland. Approximately 70% are neuroblastomas, 25% are ganglioneuroblastomas, and 5%
are ganglioneuromas (Al-Mulhim, 1998). Most of these neoplasms that metastasize to the orbit come from the adrenal gland or retroperitoneum. Metastasis to all sites occurs in 35% to 50% of all cases.
are ganglioneuromas (Al-Mulhim, 1998). Most of these neoplasms that metastasize to the orbit come from the adrenal gland or retroperitoneum. Metastasis to all sites occurs in 35% to 50% of all cases.
All tumors reported in the literature that involve the orbit are the metastatic type except for one case. This was a well-documented case of a “differentiated” neuroblastoma “primary” in the orbit, reported by Jakobiec et al. (1987). This tumor occurred in a 49-year-old woman with the tumor localized to the left orbit. Over a 12-year period, she underwent five subtotal excisions, orbital radiotherapy, and finally exenteration. However, Bullock et al. (1989) reported what was thought to be a second case of a primary orbital neuroblastoma in a 35-year-old man. On subsequent review, this case was reclassified as an orbital carcinoid tumor (Font et al., 1991).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


