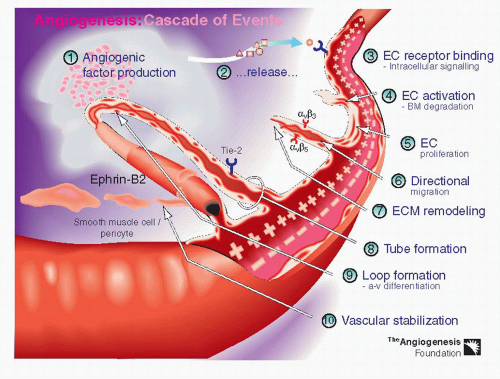
 Figure 34-1. The resulting angiogenic drive “activates” endothelial cells and precipitates a complex cascade of cellular events: proliferation of endothelial cells; migration of endothelial cells; tube formation; and ultimately formation of blood vessel loops from endothelial cell tubes. (From http://angio.org/understanding/understanding.html. © 2000 The Angiogenesis Foundation, Inc. All rights reserved.). |
(NP1, NP2) (Fig. 34-2) (6,15). VEGFR-2 appears to be the main receptor responsible for the angiogenic effects of VEGF-A. NP1 may enhance the effectiveness of VEGFR-2-mediated signaling by presenting isoform VEGF165 to VEGFR-2 (16). In humans, VEGFR-3 is thought to play a role in lymphangiogenesis (17).
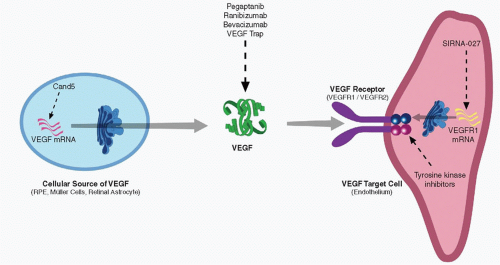 Figure 34-2. The biological effects of vascular endothelial growth factor (VEGF)-A are mediated through the VEGF-specific tyrosine-kinase receptors VEGFR-1 (Flt-1), VEGFR-2 (Flk-1), and VEGFR-3 (Flt-4), and the neuropilins (NP1, NP2). |
trials of intravitreal pegaptanib for the treatment of neovascular AMD (Table 34.1) (29). A total of 1,186 patients were randomized 1:1:1:1 to receive pegaptanib (0.3 mg, 1.0 mg, or 3.0 mg) or sham injection every 6 weeks for 48 weeks. The primary endpoint was the proportion of patients losing fewer than 15 letters of visual acuity at 54 weeks. Compared with those receiving sham injection, patients treated with 0.3 mg pegaptanib were significantly more likely to lose fewer than 15 letters of visual acuity (70% vs. 55%), more likely to maintain or improve visual acuity (33% vs. 23%), and less likely to lose 30 or more letters of visual acuity (10% vs. 22%). However, only 6% of treated patients gained 15 letters or more in visual acuity. Serious adverse events were primarily those associated with intravitreal injection: endophthalmitis, in 1.3% of patients; lens trauma, in 0.7%; and retinal detachment, in 0.6%. These events were associated with a severe loss of visual acuity in 0.1% of patients. Two-year results of the VISION study confirmed the benefit of 0.3 mg pegaptanib in reducing moderate visual acuity loss and progression to legal blindness. Advantages of pegaptanib over the prevailing standard of care, photodynamic therapy (PDT) with verteporfin, included its equal efficacy for all angiographic types of CNV and the relative ease and portability of the treatment (30). However, the treatment only demonstrated moderate improvement in visual outcome for patients and requirement of treatment at 6-week intervals versus 3-month intervals for PDT.
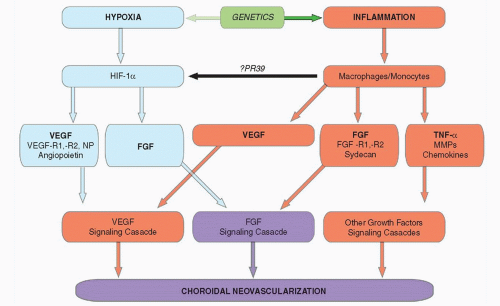 Figure 34-3. The current understanding of the pathogenesis of neovascular age-related macular degeneration (AMD). FGF, fibroblast growth factor; HIF, hypoxia inducible factor; MMP, matrix metalloproteinases; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor. |
the verteporfin group. A subgroup analysis of 12-month data from the ANCHOR study showed ranibizumab to be superior to PDT in all evaluated subgroups (37).
TABLE 34-1 PUBLISHED PHASE III CLINICAL TRIALS OF PHARMACOLOGICAL TREATMENT OF CHOROIDAL NEOVASCULARIZATION SECONDARY TO AGE-RELATED MACULAR DEGENERATION | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


