Recent technological advances now permit the study of the entire cancer genome, which can elucidate complex pathway interactions that are not apparent at the level of single genes. In this review, the authors describe innovations that have allowed for whole-exome/genome analysis of genetic and epigenetic alterations and of changes in gene expression. Studies using next-generation sequencing, array comparative genomic hybridization, methylation arrays, and gene expression profiling are reviewed, with a particular focus on findings from recent whole-exome sequencing projects. A discussion of the implications of these data on treatment and future goals for cancer genomics is included.
| BAC | Bacterial artificial chromosome |
| CDK | Cyclin-dependent kinases |
| CGH | Comparative genomic hybridization |
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| FTI | Farnesyltransferase inhibitors |
| HNSCC | Head and neck squamous cell carcinoma |
| HPV | Human papillomavirus |
| MSRE | Methylation-specific restriction enzyme |
| NGS | Next-generation sequencing |
| NICD | NOTCH1 intracellular domain |
| Rb | Retinoblastoma |
| RLGS | Restriction landmark genomic scanning |
Key points
- •
The study of head and neck squamous cell carcinoma (HNSCC) tumor development and progression is complicated by the biologic complexity and heterogeneity of the disease.
- •
Recent technological advances now permit the study of the entire cancer genome, which can elucidate complex pathway interactions that are not apparent at the level of single genes.
- •
Next-generation sequencing technology allows for the detection of base substitutions, deletions, insertions, copy number variations, and chromosomal translocations of entire exomes or genomes.
- •
Two recent whole-exome sequencing studies reported frequent mutations in TP53 , NOTCH1 , CDKN2A , PIK3CA , and HRAS in HNSCC tumors.
- •
Standard or array-based comparative genomic hybridization can detect variations in chromosomal structure with greater resolution than traditional cytogenetic techniques.
- •
Methylation and gene expression arrays can be used for gene discovery or for the identification of tumor-specific profiles that may serve as biomarkers with diagnostic or prognostic value.
Introduction
Head and neck squamous cell carcinoma (HNSCC) results from the accumulation of multiple genetic and epigenetic changes in a variety of cellular pathways. The processes of genetic alteration and selection result in the clonal expansion of those cells with the most favorable genetic aberrations, resulting in tumor development and eventual progression to invasive carcinoma.
The development and progression of cancer involves changes within multiple pathways with complex interactions. The study of the molecular underpinnings of HNSCC is further complicated by the biologic complexity of the disease. HNSCC is now known to be heterogeneous at both the histopathologic and molecular levels. The most prominent distinction is between human papillomavirus (HPV)-positive and HPV-negative tumors, but other subclasses also exist. Even within a single tumor, identification of the genes involved in carcinogenesis is hampered by tumor heterogeneity and by the interaction of tumor cells with the underlying stroma.
Cancer research has traditionally focused on the roles of individual genes in carcinogenesis. However, the study of single genes has several limitations. The process of single-gene investigation can be biased as well as labor and time intensive. Advances in technology, however, now allow for the study of the entire exome or genome. The study of the cancer genome elucidates pathways and other complex interactions that may not be apparent at the level of single genes. Whole exome/genome approaches permit the unbiased assessment of which genes and pathways have been altered. All known human genes may now be evaluated in large numbers of tumors, resulting in a more comprehensive understanding of the complex changes that occur in the formation and progression of cancer.
In this review, the authors briefly describe the recent technological advances that have allowed for whole-exome/genome analysis of genetic and epigenetic alterations as well as changes in gene expression profiles. The authors also describe some of the genes that have been implicated in HNSCC using these techniques. Finally, the authors explore implications for therapy as well as future directions for the field.
Introduction
Head and neck squamous cell carcinoma (HNSCC) results from the accumulation of multiple genetic and epigenetic changes in a variety of cellular pathways. The processes of genetic alteration and selection result in the clonal expansion of those cells with the most favorable genetic aberrations, resulting in tumor development and eventual progression to invasive carcinoma.
The development and progression of cancer involves changes within multiple pathways with complex interactions. The study of the molecular underpinnings of HNSCC is further complicated by the biologic complexity of the disease. HNSCC is now known to be heterogeneous at both the histopathologic and molecular levels. The most prominent distinction is between human papillomavirus (HPV)-positive and HPV-negative tumors, but other subclasses also exist. Even within a single tumor, identification of the genes involved in carcinogenesis is hampered by tumor heterogeneity and by the interaction of tumor cells with the underlying stroma.
Cancer research has traditionally focused on the roles of individual genes in carcinogenesis. However, the study of single genes has several limitations. The process of single-gene investigation can be biased as well as labor and time intensive. Advances in technology, however, now allow for the study of the entire exome or genome. The study of the cancer genome elucidates pathways and other complex interactions that may not be apparent at the level of single genes. Whole exome/genome approaches permit the unbiased assessment of which genes and pathways have been altered. All known human genes may now be evaluated in large numbers of tumors, resulting in a more comprehensive understanding of the complex changes that occur in the formation and progression of cancer.
In this review, the authors briefly describe the recent technological advances that have allowed for whole-exome/genome analysis of genetic and epigenetic alterations as well as changes in gene expression profiles. The authors also describe some of the genes that have been implicated in HNSCC using these techniques. Finally, the authors explore implications for therapy as well as future directions for the field.
Genetic alterations
Genetic alterations in cancer may occur in the form of small intragenic mutations, such as point mutations and insertions/deletions, or large alterations, including genomic deletions, amplifications, and chromosomal rearrangements. Whole cancer exomes/genomes can be evaluated for genetic aberrations using either next-generation sequencing (NGS) or comparative genomic hybridization (CGH) technologies.
NGS
NGS technology allows for massively parallel sequencing, producing data with greater speed and at a lower cost compared with more traditional methods. NGS instruments can process up to several million sequence reads in parallel, compared with the 96 reads produced by capillary-based instruments. In addition, template preparation, sequencing, and imaging steps for NGS platforms are highly automated and streamlined, requiring less time and additional equipment than high-throughput capillary-based sequencing systems.
NGS has several other advantages over capillary-based methods, such as Sanger sequencing. Sanger sequencing has generally been limited to the analysis of either single genes or select hot-spot regions within the genome. In contrast, NGS allows the detection of base substitutions, deletions, insertions, copy number variations, and chromosomal translocations. NGS technologies have increased sequencing rates by several orders of magnitude and significantly reduced the cost per base, making it possible to sequence all known genes in multiple tumors of a given cancer type or in matched tumor and normal tissues.
Sequencing of either whole exomes or genomes can be performed using NGS platforms. Protein-coding regions constitute only about 1% of the human genome but are thought to account for 85% of mutations resulting in disease. Because it targets that part of the genome enriched for causative genes, whole-exome sequencing is efficient, affordable, and allows many more samples to be sequenced. In addition, because of the exome enrichment and higher base coverage, whole-exome platforms are currently more sensitive than whole-genome technologies for the detection of variants within coding regions. However, whole-exome sequencing cannot identify variants in noncoding regions or genomic structural variations, whereas whole-genome sequencing can identify both. Although greater genomic coverage may be useful, whole-genome sequencing generates vast amounts of data of yet unknown functional and clinical significance.
Recently, 2 studies were published in which whole-exome sequencing was performed in a total of 106 primary HNSCC tumors with matched normal DNA. Mutations were confirmed in several genes, including in TP53 , CDKN2A , FAT1 , PTEN , HRAS , PIK3CA, and EGFR , that had been previously implicated in HNSCC. Both studies also identified mutations in NOTCH1 , which had never previously been associated with HNSCC ( Fig. 1 ).
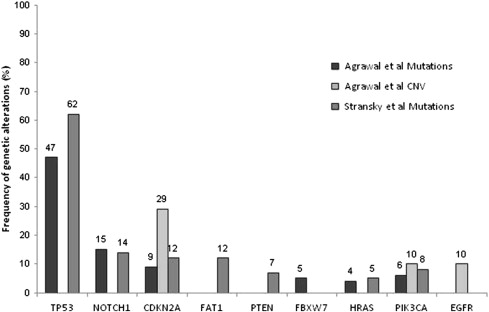
The authors briefly review the most commonly mutated genes in HNSCC, as identified in these two studies ( Table 1 ).
| Gene Symbol | Gene Name | Chromosomal Location | Gene Function | Mutation Rate | Copy Number Variation | References |
|---|---|---|---|---|---|---|
| Tumor Suppressor Genes | ||||||
| TP53 | Tumor protein p53 | 17p13.1 | Tumor suppressor that assists in cell cycle arrest, DNA damage repair, apoptosis, and senescence. | 40%–62% | N/A | |
| NOTCH1 | Notch1 | 9p34.3 | Tumor suppressor or oncogene (tissue dependent) that is important in regulation of cell differentiation, lineage commitment, and embryonic development. | 14%–15% | N/A | |
| CDKN2A | Cyclin-dependent kinase inhibitor 2A | 9p21.3 | Tumor suppressor that is important in cell cycle regulation. | 9%–12% | 29% | |
| FAT1 | FAT tumor suppressor homolog 1 ( Drosophila ) | 4q35.2 | Tumor suppressor that is a member of cadherin family and involved in cell adhesion, migration, and invasion. | 12%–80% | N/A | |
| PTEN | Phosphatase and tensin homolog | 10q23.3 | Tumor suppressor that is a negative regulator of PI3K. | 7%–10% | N/A | |
| FBXW7 | F-box and WD repeat domain-containing protein 7 | 4q31.3 | Tumor suppressor that is a member of the F-box protein family and component of the ubiquitin ligase complex that can mediate NOTCH1, cyclin E and c-myc degradation. | 5% | N/A | |
| Oncogenes | ||||||
| HRAS | Harvey rat sarcoma viral oncogene homolog | 11p15.5 | Oncogene; GTPase that is important in promoting cell proliferation, differentiation, and survival through downstream effector pathways. | 4%–35% | N/A | |
| PIK3CA | Phosphoinositide-3-kinase catalytic alpha polypeptide | 3q26.32 | Oncogene that is a catalytic subunit of PI3K, a target of Ras activation, and promotes cell growth, survival, and cytoskeleton reorganization. | 6%–8% | 10% | |
| EGFR | Epidermal growth factor receptor | 7p12 | Oncogene that is a receptor tyrosine kinase in the ErbB family and involved in cell proliferation, apoptosis, invasion, angiogenesis, and metastasis. | N/A | 10% | |
Tumor suppressor genes
TP53
The NGS studies confirmed the well-established role of TP53 , a tumor suppressor gene on chromosome 17p12, in HNSCC. Mutated in approximately half of all HNSCC tumors, TP53 is the most commonly mutated gene in HNSCC. Functional loss of p53 has been demonstrated in many human cancers and plays a critical role in malignant transformation. In fact, in the carcinogenesis of HNSCC, mutations in TP53 occur early. TP53 mutations are present in dysplastic premalignant lesions of the oral cavity, and the prevalence of these mutations increases with histopathologic progression of the tumor from dysplasia to invasive carcinoma.
Under normal circumstances, in response to DNA damage, p53 accumulates within the nucleus and causes cell cycle arrest via transcriptional induction of downstream effectors. If DNA repair is unsuccessful, p53 triggers apoptosis or senescence. However, cells harboring mutations in TP53 will not undergo cell cycle arrest, apoptosis, or senescence. The p53-deficient cells can replicate in the presence of damaged DNA and accumulate additional genetic mutations, leading to unchecked cell division and tumor formation and progression.
TP53 mutations in HNSCC have been associated with poor clinical outcomes as well as poor response to treatment. A large prospective multicenter trial including 420 patients found that mutations disruptive to the DNA-binding domain of p53 decreased overall survival by 1.7 times compared with patients without disruptive mutations. Poor tumor response to radiation or chemotherapy has also been associated with mutations in TP53 . Alterations of p53 by mutation, deletion, or other mechanisms of inactivation have been found in 95% of HNSCC tumors refractory to radiation. The risks of locoregional recurrence and death after either primary or postoperative radiation therapy have both been found to be significantly greater for patients with mutations in TP53. TP53 mutations have also been associated with poor response to cisplatin and fluorouracil. In a prospective study of 106 patients with HNSCC, TP53 mutations were more frequent in patients who did not respond to cisplatin and fluorouracil than in patients who did respond. TP53 mutation status was found to be an independent predictor of response to chemotherapy.
NOTCH1
NOTCH1 is the second most commonly mutated gene in HNSCC, with a mutation rate of 14% to 15%. NOTCH1 is important in regulating normal cell differentiation, lineage commitment, and embryonic development. It seems to function as a tumor suppressor gene in HNSCC based on the position and characteristics of the mutations and the inactivation of both alleles. NOTCH1 is thought to act as a tumor suppressor gene in several other human cancers, including cutaneous squamous cell carcinoma (SCC), lung SCC, and chronic myelomonocytic leukemia, although it seems to function as an oncogene in other leukemias.
The NOTCH1 protein is a transmembrane ligand receptor with intracellular and extracellular domains. On ligand binding, the NOTCH1 intracellular domain (NICD) is cleaved and translocates to the nucleus. In the nucleus, the NICD activates transcription by binding to CBF1 in the presence of coactivators from the Mastermindlike family. Downstream target genes of NOTCH1 signaling are crucial for cell differentiation and normal embryonic development.
Activating and loss-of-function mutations preferentially occur in different regions of the NOTCH1 gene ( Fig. 2 ). Most NOTCH1 mutations observed in HNSCC affect the epidermal growth factor (EGF)-like ligand-binding domain and are thought to lead to loss of function. Inactivating mutations in these regions of the gene have also been reported in skin and lung SCC. In contrast, mutations of the intracellular proline, glutamic acid, serine/threonine-rich motifs regulatory domain or the extracellular heterodimer domain are thought to result in constitutive activation of NOTCH1 signaling.
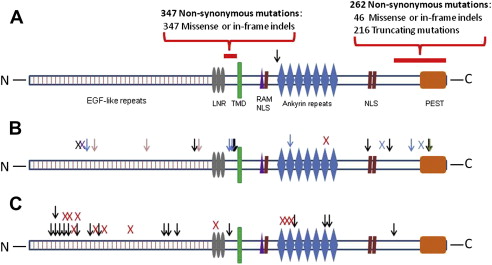
Mutations in the gene FBXW7 have also been identified in 5% of HNSCC specimens. FBXW7 is a member of the F-box protein family and is a component of the ubiquitin ligase complex that can mediate NOTCH1 degradation. FBXW7 mutations could, therefore, also affect the NOTCH1 pathway, although FBXW7 is also known to target other oncogenic pathways, such as cyclin E and c-myc.
Cyclin-dependent kinase inhibitor 2A (CDKN2A)
Alterations of CDKN2A/ p16 INK4A , a tumor suppressor gene located on chromosome 9p21, have long been recognized in HNSCC. In the NGS studies, CDKN2A mutations were identified in 9% to 12% of all tumors. Gene copy number analyses also revealed frequent loss of heterozygosity and deletions of CDKN2A. In addition to deletions and point mutations, CDKN2A is also inactivated by methylation of the 5′ CpG region.
The protein product of CDKN2A , p16, plays a critical role in cell cycle regulation via its interaction with the retinoblastoma (Rb) tumor suppressor. The p16 protein inhibits cyclin-dependent kinases (CDK) 4 and 6, which are in turn necessary for the phosphorylation of Rb. Hypophosphorylated Rb induces G1 arrest of the cell cycle.
Alterations of CDKN2A , although they are common events in early HNSCC development, alone are likely insufficient to drive tumorigenesis. This point is supported by the fact that CDKN2A mutations have been reported in benign epithelial lesions with low potential for malignant transformation.
FAT1
FAT1 is a tumor suppressor gene in the cadherin family of integral membrane proteins and is involved in cell adhesion, migration, and invasion. It was found to have a 12% incidence of mutation. In previous studies, homozygous deletions in FAT1 were identified in most oral SCCs.
PTEN
PTEN is a negative regulator of PI3K, and its loss results in activation of the PI3K/Akt pathway, which is discussed later. Inactivating mutations were found in PTEN in 7% of tumors, consistent with previous reports of mutation rates as high as 10%.
Oncogenes
HRAS
The true incidence of Ras mutations in HNSCC has been unclear. The reported frequency of mutations in the Ras family gene HRAS in HNSCC has varied from 0% in Western populations to as high as 35% in Indian populations. It is thought that these differences in mutation frequencies are related to the use of tobacco chewing and betel quid habits in Indian and other Asian countries, although it may also reflect underlying genetic variations between ethnic groups. Both of the NGS projects confirmed the presence of HRAS mutations in HNSCC, with a frequency of 4% to 5%.
Ras proteins are GTPases that function as signaling switches by alternating between the guanosine triphosphate–bound active state and the guanosine diphosphate–bound inactive state. Ras downstream effector pathways include Raf. Activated Raf phosphorylates MEK, which in turn activates ERK. The Raf/MEK/ERK pathway is involved in the regulation of cell proliferation, differentiation, and survival.
PIK3CA
Mutations were identified in the oncogene PIK3CA in 6% to 8% of tumors. Previous studies have found rates of PIK3CA mutation as high as 20%. PIK3CA, which encodes the catalytic subunit p110alpha of the PI3K heterodimer, activates the AKT/mTOR pathway and promotes cell growth, cell survival, transformation, and drug resistance.
EGF receptor
The EGF receptor (EGFR) is a receptor tyrosine kinase that belongs to the ErbB family of cell surface receptors and is involved in cellular proliferation, apoptosis, invasion, angiogenesis, and metastasis via the MAPK, AKT, ERK and JAK/STAT pathways. Focal amplification of 7p, which contains the EGFR gene, was found in approximately 10% of samples by copy number analysis. Dysfunction of the EGFR pathway has been described in 80% to 90% of HNSCC, with overexpression of EGFR being the most common cause of dysregulation.
Comparative Genomic Hybridization
Variations in chromosomal structure, including inversions, deletions, translocations, or gains or losses of entire chromosomal segments, are common in the development and progression of HNSCC. Resultant DNA copy number alterations may change gene expression and function, resulting in tumor development.
The most commonly described chromosomal aberrations in HNSCC include the following :
- •
Gains in 1q, 3q, 5p, 7p, 8q, 9q, 11q, 14q, and 18p
- •
Losses in 3p, 4p, 4q, 5q, 8p, 9p, 10p, 11q, 13q, 17p, 18q, and 21q
Many of these genomic gains and losses were originally detected using loss of heterozygosity analysis or traditional cytogenetic techniques, such as karyotype analysis. However, karyotype analysis is technically challenging to perform in solid tumors and, furthermore, does not allow detection of submicroscopic losses or rearrangements.
CGH permits the efficient analysis of the entire genome for DNA copy number variation
However, the minimum size of a detectable segment in standard CGH is 3 to 5 Mb, which limits the detection of smaller alterations and makes identification of specific candidate genes difficult. More recently, microarray-based assays, or array CGH, have been developed to overcome the pitfalls of karyotype and standard CGH-based analyses. Array CGH, which uses bacterial artificial chromosome (BAC) arrays, allows for the high-throughput analysis of DNA copy number variations throughout the whole genome and allows high-resolution mapping of these changes directly onto genomic sequence. The resolution of array CGH platforms continues to improve to submegabase levels, so that variations ranging from gene-size aberrations to entire chromosomal arms may now be detected. Of note, whole-genome analysis of copy number variation can also be assessed using newer single-nucleotide polymorphism array platforms, which have the additional advantage of being able to detect the loss of heterozygosity. However, to date, most whole-genome studies of copy number variation in HNSCC have used BAC-based array CGH ( Table 2 ).
| Author, Year | Methodology | Cohort | Findings |
|---|---|---|---|
| Snijders et al, 2005 | In-house HumArray2.0 (UCSF) | 89 oral SCC | Identified 9 recurrent amplified regions <3 Mb, which contained genes involved in integrin signaling, adhesion, migration, survival, Hedgehog, and Notch pathways, which were amplified and overexpressed. |
| Garnis et al, 2004 | In-house BAC array | 22 oral SCC | Identified 5.3 Mb region of common amplification at 8q22 containing 16 known genes. Gene expression analysis revealed overexpression of LRP12, a novel putative oncogene. |
| Smeets et al, 2006 | In-house BAC array | 12 oral SCC, 12 oropharynx SCC | Compared HPV-positive and HPV-negative tumors. Four regions of gains or losses were unique to HPV-negative tumors. Seven regions of gains or losses were altered at high frequency in both tumor subsets. |
| Ashman et al, 2003 | Standard CGH | 10 oral or oropharynx SCC, 35 other HNSCC | Gain of 3q25–q27 and loss of 22q were associated with reduced disease-specific survival. Gains of 17q and 20q, loss of 19p and 22q, and amplification of 11q13 were associated with reduced disease-free survival. |
| Ambatipudi et al, 2011 | Human Genome CGH Microarray 105K (Agilent) | 60 oral SCC | Gain of 11q22.1–q22.2 and losses of 17p13.3 and 11q23–q25 were associated with earlier loco-regional recurrence and shorter overall survival. |
| Uchida et al, 2011 | MacArray Karyo 4K (Macrogen) | 50 oral SCC | Loss of a 0.2 Mb region at 3p26.3 was associated with reduced disease-specific survival. |
| Sugahara et al, 2011 | Human Genome CGH Microarray 44 K (Agilent) | 54 oral SCC (22 with cervical LN metastasis, 32 without cervical LN metastasis) | Compared tumors that had or had not metastasized to the cervical lymph nodes. Two distinct regions of amplification at 11q13 were associated with cervical metastasis. |
| van den Broek et al, 2007 | Standard CGH | 34 oral or oropharynx SCC, 6 other HNSCC | Compared chemoradiotherapy-sensitive and resistant tumors. Gains of 3q11-q13, 3q21-q26.1, and 6q22-q27 and losses of 3p11-pter and 4p11-pter were associated with chemoradiotherapy resistance. |
Array CGH has been used in several studies to identify candidate pathways and genes in HNSCC tumorigenesis
Sparano and colleagues identified 22 amplified and 17 deleted genes in at least 25% of oral SCC. Snijders and colleagues used array CGH to identify several common regions of chromosomal amplification containing genes involved in integrin signaling, adhesion, migration, and survival pathways in oral SCC. They also found dysregulation of the Hedgehog and Notch pathways. In another study, array CGH was used to identify a novel gene possibly involved in oral SCC tumorigenesis. Two neighboring regions on chromosome 8q were found to be amplified, one of which harbored a novel putative oncogene, LRP12 . Overexpression of LRP12 , but not of two flanking genes, was then confirmed using reverse transcription–polymerase chain reaction (RT-PCR).
Array CGH has been used to attempt to delineate differences in the molecular and clinical characteristics of tumors
Smeets and colleagues used array CGH to compare HPV-positive and HPV-negative tumors. The two subsets of tumors were found to harbor different genomic gains and losses, although both tumor types shared some genetic changes. This finding provides evidence of differences in the genetic alterations observed in HPV-positive compared with HPV-negative tumors.
Associations between clinical outcomes and specific structural variations have also been identified. A study using standard CGH in HNSCC identified significant associations between a gain of 3q25 to 3q27 and a loss of 22q with reduced disease-specific survival and a gain of 17q and 20q and a deletion of 19p and 22q with reduced disease-free survival. A more recent study conducted array CGH in advanced-stage oral SCC and found that a gain of region 11q22.1-q22.2 and losses of 17p13.3 and 11q23-q25 were significantly associated with earlier locoregional recurrence and shorter overall survival. Uchida and colleagues found that a loss of 3p26.3 was significantly associated with poorer disease-specific survival. Another study demonstrated an association between amplification at 2 distinct regions of 11q13 and metastases to the cervical lymph nodes.
CGH has also been used to predict differences in response to treatment
Although such studies have not yet been performed using array-based approaches. Akervall and colleagues demonstrated that cisplatin-resistant cell lines had higher rates of chromosomal gains and losses compared with cisplatin-sensitive cell lines. Another study used standard CGH to compare chemoradiotherapy-resistant primary HNSCC tumors with sensitive tumors. Both groups had similar total numbers of genetic changes, but high-level DNA amplifications were more frequent in the resistant tumors. Several specific chromosomal gains and losses were significantly associated with resistance to treatment. Such genomic profiles may be useful as predictors of resistance to chemoradiotherapy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


