Purpose
To investigate the genetic cause and perform a comprehensive clinical analysis of a Danish family with autosomal recessive bestrophinopathy; to investigate whether Bestrophin may be expressed in normal human retina.
Design
Retrospective clinical and molecular genetic analysis and immunohistochemical observational study.
Methods
setting : National referral center. participants : A family with 5 individuals and biallelic BEST1 mutations, and enucleated eyes from 2 individuals with nonaffected retinas. observation procedures : Molecular genetic analysis included sequencing of BEST1 and co-segregation analysis. Clinical investigations included electro-oculography, full-field electroretinography, multifocal electroretinography, spectral-domain optical coherence tomography, and fundus autofluorescence imaging. Immunohistochemical analysis was performed. main outcome measures : BEST1 mutations, imaging findings, electroretinography amplitudes, and implicit times.
Results
The index case was compound heterozygous for p.A195V and a novel 15 base pair deletion leading to p.Q238L. The index case at age 10 demonstrated multifocal vitelliform changes that were hyperautofluorescent, cystoid macular edema in the inner nuclear layer, no light rise in the electro-oculography, and a reduced central but preserved peripheral retinal function by multifocal electroretinography. Full-field electroretinography demonstrated a reduced rod response and inner retina dysfunction. Retinal structure was normal in all 3 family members who carried a sequence change in BEST1 . Electro-oculography light peak was reduced in both the mother and sister (heterozygous for p.Q238L). Immunohistochemistry could not confirm the presence of Bestrophin in normal human retina.
Conclusions
Because of a relatively well preserved retinal function, autosomal recessive bestrophinopathy may be a suitable first candidate, among the BEST1 -related ocular conditions, for gene replacement therapy.
Different mutations in the BEST1 gene can cause a variety of ocular phenotypes, ranging from isolated vitelliform macular degeneration (owing to disturbed function of the retinal pigment epithelium), which manifests as a reduced light peak in the electro-oculogram as in Best disease, to widespread retinal degeneration as in autosomal recessive bestrophinopathy, and further to widespread ocular manifestations as in autosomal dominant vitreoretinochoroidopathy. The phenotypes described as autosomal dominant and autosomal recessive retinitis pigmentosa resemble those of autosomal dominant vitreoretinochoroidopathy and autosomal recessive bestrophinopathy, respectively. Although a large number of patients with BEST1 mutations have been recently reported with different phenotypes, no clear genotype-phenotype correlation can be obtained for BEST1 -associated ocular diseases. This can be partially explained by the considerable variability in disease expressivity among patients who carry the same BEST1 disease–causing mutation.
Autosomal recessive bestrophinopathy seems to be intermediate in severity among the bestrophinopathies and is characterized by biallelic mutations in BEST1 , the absence of a light rise in the electro-oculography, reduced full-field electroretinogram, pigment irregularities, and vitelliform changes in the posterior pole of the fundus and may include cystoid macular edema and angle-closure glaucoma. However, other biallelic mutations may manifest as classical Best disease with no signs of generalized involvement of the retina except for a reduced electro-oculography light rise. Furthermore, angle-closure glaucoma was also recently described in classical Best disease.
The electro-oculogram measures the resting potential across the retinal pigment epithelium. The clinical hallmark of Best disease and other BEST1 -associated retinopathies is a reduction of the electro-oculography light rise, which indicates a disturbed function of the retinal pigment epithelium. This reduction is consistently pronounced in autosomal recessive bestrophinopathy, where the light rise in the electro-oculogram may be absent or nearly absent. The gene product of BEST1 was previously postulated to act as an calcium-sensitive chloride channel in the basolateral retinal pigment epithelium; however, a recent study showed that bestrophin is localized in the endoplasmic reticulum membrane, close to the cell membrane. In vitro studies, in which cells were transfected with mutant and wild-type bestrophin and chloride currents were measured, have shown that certain recessive mutations associated with autosomal recessive bestrophinopathy (mutations p.R141H and p.P152A) resulted in diminished chloride currents compared to wild-type. Subsequent cotransfection with wild-type bestrophin resulted in currents that were not different from cells transfected with wild-type protein alone, consistent with the recessive nature of the disease. By contrast, cotransfection with wild-type protein did not restore chloride currents in cells that were previously transfected with mutated bestrophin owing to dominant mutations (p.W93C and p.R218C). Thus, lack of channel conductance may underlie or contribute to the reduction or absence of a light rise in the electro-oculogram and may be a pathogenic mechanism of particular relevance in autosomal recessive bestrophinopathy.
In this study we present a detailed clinical and genetic investigation of a family with biallelic mutations in BEST1 , in an attempt to gain additional information on the mechanism by which different BEST1 mutations cause a variety of ocular phenotypes. The biallelic form of BEST1 -associated retinopathy is of particular interest because an animal model exists, namely canine multifocal retinopathy, for which gene therapy has shown sustained therapeutic effect (Guziewicz K, et al. IOVS 2013;54:ARVO E-Abstract 5965). Although the autosomal recessive bestrophinopathy phenotype has been presented in many papers since its original description by Burgess and associates in 2008, we herein present and evaluate several novel molecular genetic and clinical aspects in members of a family with biallelic mutations in BEST1 . These new findings and investigations include: (1) the presence of 3 sequence alterations in BEST1 , 1 of which is a novel deletion of 15 nucleotides; (2) data on choroidal thickness and anterior chamber angle; and (3) evaluation of the luminance-response series in the full-field electroretinogram. Finally, as structural and functional data indicated involvement of the inner retina in autosomal recessive bestrophinopathy, we performed immunohistochemistry to investigate whether Bestrophin may be expressed in the normal human retina.
Patients and Methods
This retrospective study included a 10-year-old index case with vitelliform maculopathy and 5 first-degree relatives ( Figure 1 ). In addition, for immunohistochemical studies we used 2 adult human eyes, which had been enucleated because of a malignant tumor and ocular trauma. Prior to donation of a blood sample for DNA analysis, informed consent was obtained from all patients and family members who participated in this study. Ethical permission for DNA analysis and clinical investigation in patients with hereditary retinal degeneration was obtained from the central ethical review board at Lund University, Sweden. Institutional review board approval is not granted for retrospective studies in Denmark. In Denmark, institutional review boards do not provide waivers. This was not a systematic evaluation of a treatment or a device. The tenets of the Declaration of Helsinki were followed, as were all federal or state laws in the countries (Israel, Denmark, Sweden) involved in the study.
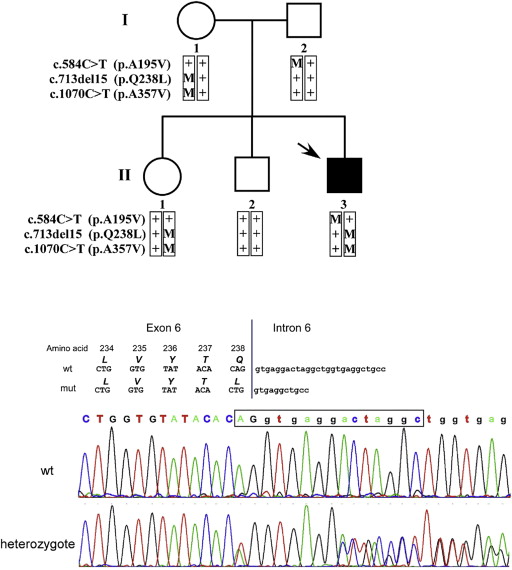
Molecular Genetic Methods
Genomic DNA was extracted from peripheral blood of all family members using standard procedures. Molecular genetic analysis included Sanger sequencing of the BEST1 coding region and intron-exon junctions and a co-segregation analysis in the studied family. Primers for exons 1-3 and 5-11 of BEST1 were previously reported. Primers for exon 4 were: forward 5′-AGAAAGCTGGAGGAGCCGA-3′ and reverse 5′-TCCACCCATCTTCCATTCCTGC-3′. The predicted effect of a sequence change on splicing was evaluated using the splice-site prediction by neural network platform ( http://www.fruitfly.org/seq_tools/splice.html ; accessed September 3, 2013). The possible pathogenicity of missense changes was evaluated using PolyPhen-2 ( http://genetics.bwh.harvard.edu/pph2/ ; accessed September 3, 2013), MutationTaster ( http://www.mutationtaster.org ; accessed September 3, 2013), and SIFT ( http://sift.jcvi.org ; accessed September 3, 2013).
Clinical Investigations
Investigations included standard clinical examination, multifocal electroretinography, fundus autofluorescence imaging, and spectral-domain optical coherence tomography, full-field electroretinography, and electro-oculography as described previously, with some modification as follows.
Electrophysiologic examinations were conducted at the Kennedy Center, Denmark, according to the standards given by the International Society of Clinical Electrophysiology in Vision, with 1 exception: Dawson-Trick-Litzkow electrodes were used instead of Burian-Allen contact lens electrodes because of contact lens intolerance in the index case and the choice of using the same method in all family members. For both electro-oculography and full-field electroretinography, we used a Viking 5.0 Ganzfeld dome (Nicolet Biomedical Instruments, Madison, Wisconsin, USA), with a light-emitting diode for light stimulation. The Ganzfeld dome was equipped with an infrared camera to monitor the position of the eye during the examination.
In addition, the dark-adapted full-field electroretinography was extended to include an intensity response series outside the standard given by the International Society of Clinical Electrophysiology in Vision, with 7 stimulus intensities between −1.5 and +1.5 log units with a 0.5 log unit interval, where the standard flash luminance of 3.0 candela-seconds per square meter corresponds to 1.0 log units. Each response represented an average of at least 2 measurements of comparable size and shape, if possible.
In both full-field electroretinography and multifocal electroretinography, the reference electrode was positioned inferior to the lateral canthus and the ground electrode on the earlobe. The examined eye was locally anesthetized with oxybuprocaine 0.4% prior to positioning the Dawson-Trick-Litzkow electrode.
Multifocal electroretinography was performed using VERIS 4.0 (Electro-Diagnostic Imaging, San Diego, California, USA) with charge-coupled device camera. The stimulus pattern of multifocal electroretinography consisted of 103 hexagons. The multifocal electroretinography examination was subdivided into 16 segments of 30 seconds. A short break of 2-5 seconds was given after each segment. We used 17% averaging from neighboring hexagons and 2 iterations of artefact removal when calculating multifocal electroretinography ring averages and presenting trace plots.
Anterior segment optical coherence tomography (Visante OCT; Carl Zeiss Meditec, Jena, Germany) and/or Scheimpflug Pentacam (Oculus Optikgeräte GmbH, Wetzlar, Germany) was performed to visualize and measure the anterior chamber angle and the subfoveal choroidal thickness was examined using spectral-domain optical coherence tomography and enhanced depth imaging and measured as described previously.
Immunohistochemistry
Eyes from 2 unrelated individuals with no known BEST1 -related disease were enucleated because of a malignant choroidal melanoma or ocular trauma. The malignant tumor was located well away from the sampled tissue. Samples were fixed in 4% paraformaldehyde, embedded on paraplast, and cut. After deparaffinization and rehydration, sections were incubated in a decloaking chamber (Biocare Medical, Concord, California, USA) with 10 mM citrate buffer (pH 6.0) at 125 C for 10 minutes. Hereafter they were rinsed several times with phosphate-buffered saline; blocked with phosphate-buffered saline containing 1% bovine serum albumin, 0.1% triton-X100, and 10% normal donkey serum; and subsequently incubated overnight at 4 C with anti-Bestrophin 1 primary antibody (mouse monoclonal, 1:50; Novus Biologicals, Littleton, Colorado, USA). After washing in phosphate-buffered saline, the secondary antibody was applied for 1 hour (DyLight 549 AffiniPure Donkey Anti-Mouse IgG, 1:250; Jackson ImmunoResearch Laboratories, Inc, West Grove, Pennsylvania, USA). Nuclei were counterstained with 4,6-diamidino-2-phenylindole–containing mounting medium (Vector Laboratories, Burlingame, California, USA). To determine the specificity of the antigen-antibody reaction, corresponding negative controls with the secondary antibody alone were performed. Observation and photography were performed using a fluorescent microscope (Olympus BX41, Tokyo, Japan) equipped with a DP70 digital camera.
Results
Mutation Analysis
Sequencing analysis of BEST1 in the index case (a 10-year-old boy, II:3, Figure 1 ) revealed 3 heterozygous sequence changes ( Supplementary Table , available at AJO.com ). Two sequence changes are missense (eg, affect only 1 amino acid) and were previously reported by others: the p.A195V change (alanine at position 195 is replaced by valine) was described previously in Best disease, in autosomal recessive bestrophinopathy, and in multifocal Best disease; and the p.A357V change (alanine at position 357 is replaced by valine) was reported in patients with Best disease. In addition, we identified a novel deletion of 15 nucleotides (termed c.713del15; the deletion includes the last 2 nucleotides of exon 6 and the first 13 nucleotides of intron 6; Figure 1 ). Although deletions that involve the exon-intron junction are likely to affect mRNA splicing, the deletion we identified is unique owing to sequence similarities between the mutated and wild-type sequence and is thus not predicted to affect the normal splicing of exon 6 (as predicted by splicing prediction tools), but instead results in a novel missense change (p.Q238L). Based on the reported allele frequency and the pathogenicity prediction for each sequence change ( Supplementary Table ), we predict that p.A195V and p.Q238L are disease-causing mutations, whereas p.A357V is a rare nonpathogenic variant. We examined BEST1 alleles in all 5 family members and were able to determine the segregating alleles in the family ( Figure 1 ). The index case is compound heterozygous for p.A195V (which was inherited from the father) and p.Q238L (in cis with p.A357V, inherited from the mother). None of the 2 siblings inherited this combination of alleles; the sister inherited the p.Q238L-p.A357V allele and the brother inherited 2 normal copies of the BEST1 gene ( Figure 1 ).
Clinical Findings
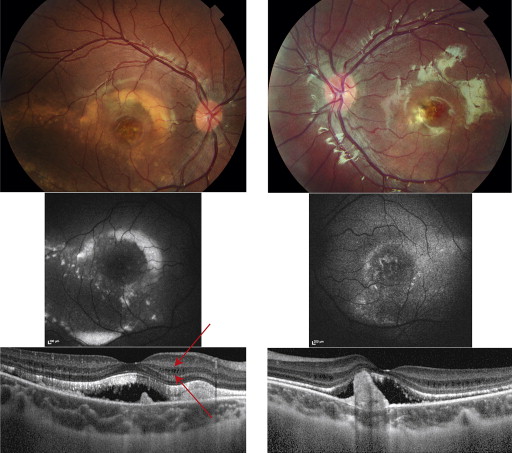
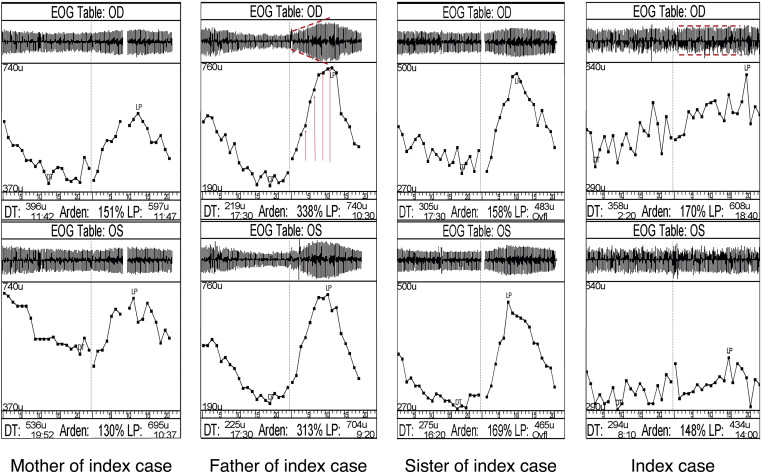
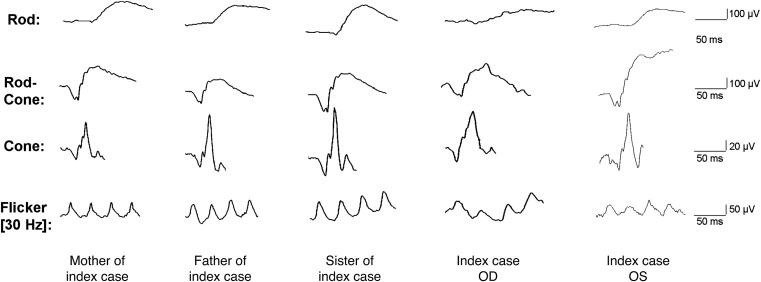
The index case at the age of 10 years had widespread vitelliform changes in the posterior pole ( Figure 2 , Top) that were hyperautofluorescent ( Figure 2 , Middle), indicating the presence and accumulation of retinoid derivatives such as lipofuscin. Photoreceptor outer segments were elongated in a precipitate-like manner and focal mounds were present on the Bruch membrane, these structures being separated by subretinal fluid. Cystoid macular edema localized in the inner nuclear layer ( Figure 2 , Bottom; arrows indicate the limits of the inner nuclear layer). Multifocal electroretinography demonstrated a reduced central but preserved paracentral retinal function in the right eye, as demonstrated by the analysis of ring ratios, given as the ratio of the responses from the center (rings 1-2) vs those from the periphery (rings 3-6) ( Table 1 and Figure 3 ). Analysis of absolute amplitudes in these rings demonstrated a more pronounced amplitude reduction throughout the field in the left eye, as compared to the right eye. There was no consistent light rise in the electro-oculography ( Table 1 and Figure 4 ).
| Patient | Sex (Age) | BEST1 Genotype | Multifocal Electroretinography Amplitude Ring Averages for P1 Wave Ring 1-2, 3-6, Ring Ratio b (nV/deg 2 ) | Electrooculogram Arden Ratio (OD, OS) | Visual Acuity OD, OS (Refraction) | Subfoveal Choroidal Thickness OD, OS (μm) | Anterior Chamber Depth OD,OS (mm) | Anterior Chamber Angle OD Temporal, Nasal (Degrees) |
|---|---|---|---|---|---|---|---|---|
| I:1 | F (51) | +/Q238L and A357V | 59.4, 28.1, 2.1 (OD) | 1.51, 1.30 | 1.3, 1.3 (−1) | 512, 464 | 2.33, 2.34 | 26.1, 20.0 |
| I:2 | M (51) | +/A195V | 62.1, 26.6, 2.3 (OD) | 3.38, 3.13 | 1.3, 1.3 (0) | 249, 288 | 3.65, 3.60 | 41.7, 39.2 |
| II:1 | F (27) | +/Q238L and A357V | 56.0, 32.4, 1.7 (OD) | 1.58, 1.69 | 1.3, 1.3 (0) | Not analyzed | 3.65, 3.72 | 31.5, 28.2 |
| II:2 | M (23) | +/+ | Not analyzed | Not analyzed | 1.3, 1.8 (−2.5) | Not analyzed | Not analyzed | Not analyzed |
| II:3 | M (10) | A195V/Q238L and A357V | 49.9, 37.5, 1.3 (OD) 23.8, 12.3, 1.9 (OS) | 1.70, 1.48 | 0.9, 0.28 (+1) | 281, 219 | 3.48, 3.52 | 41.1, 44.6 |
| Normal median (range) a | 71.3 (63.6-95.0), 38.3 (20.6-46.3), 2.14 (1.60, 3.36) | 342 (73-691) |
a Normal data were obtained with a Dawson-Trick-Litzkow electrode at the Kennedy Center, based on the investigation of 10 eyes of 8 controls, age range 30-60 years.
b Multifocal electroretinography ring ratio is given as ring 1-2 amplitude average divided by ring 3-6 amplitude average.
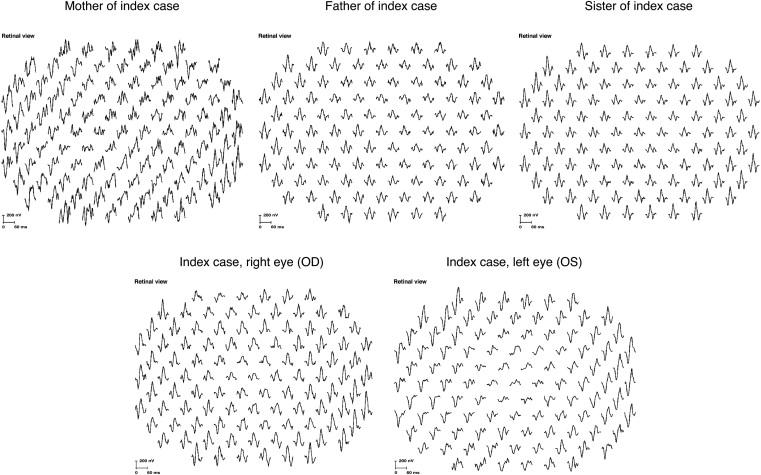
Full-field electroretinography indicated reduced rod-driven response in the index case compared to other family members, while the cone-driven responses were better preserved, except for a delay in 30 Hz flicker implicit time ( Figure 5 and Table 2 ). A further evaluation of retinal function with an intensity response series in the dark-adapted full-field electroretinography demonstrated lower amplitude in the index case only at the dimmest stimulus luminance, corresponding to the rod-dominated response; however, with increasing stimulus luminance, and thus increasing contribution from cones, the response was comparable to that of other family members (right eye of index case) or even exceeded that of other family members (left eye of index case) ( Table 2 , Figures 6 and 7 ).
| Patient (Eye) | Sex (Age) | BEST1 Mutation | Full-Field Electroretinography | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rod B Wave | Rod-Cone A Wave | Rod-Cone B Wave | Cone B Wave | 30 Hz Flicker | ||||||||
| Amplitude | Implicit Time | Amplitude | Implicit Time | Amplitude | Implicit Time | Amplitude | Implicit Time | Amplitude | Implicit Time | |||
| I:1 (OD) | F (51) | +/Q238L and A357V | 266 | 86 | 176 | 22 | 427 | 55 | 92 | 32 | 94 | 28 |
| I:2 (OD) | M (51) | +/A195V | 212 | 81 | 160 | 22 | 310 | 56 | 131 | 33 | 125 | 29 |
| II:1 (OD) | F (27) | +/Q238L and A357V | 378 | 86 | 220 | 22 | 491 | 56 | 170 | 30 | 124 | 27 |
| II:2 (N/A) | M (23) | +/+ | N/A | |||||||||
| II:3 (OD) | M (10) | A195V/Q238L and A357V | 195 | 101 | 160 | 23 | 424 | 57 | 124 | 33 | 80.8 | 30 |
| II:3 (OS) | M (10) | A195V/Q238L and A357V | 200 | 79.0 | 158 | 23 | 646 | 62 | 143 | 32 | 98 | 31.5 |
| Normal median, (range) a | 263 (193-405) | 81.5 (64.5-93) | 229 (187-269) | 20.5 (15.5-23.0) | 479 (322-501) | 42.5 (35.0-59.0) | 157 (105-168) | 42.3 (24.5-62.5) | 135 (83.7-165) | 27.0 (24.5-29.0) | ||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


