This article discusses the epidemiology, presentation, and diagnostic work-up of nonparaganglioma jugular foramen tumors, and the management options and predicted outcomes. Paragangliomas are the most common jugular foramen tumors, but other nonparagangliomas are important to consider in a differential for jugular foramen tumors. This article specifically focuses on jugular foramen schwannomas, meningiomas, metastatic disease, and regional pathologies that may extend to the jugular foramen, such as endolymphatic sac tumors, chordomas, and chondrosarcomas. Operative approaches to these tumors are also reviewed.
Key points
- •
Nonparaganglioma jugular foramen tumors are rare tumors and include schwannomas, meningiomas, endolymphatic sac tumors, chordomas, and chondrosarcomas, and metastatic disease spread to the jugular foramen.
- •
The clinical presentation of any jugular foramen tumor may include symptoms related to neuropathies of the facial, vestibulocochlear, or lower cranial nerves; pulsatile tinnitus; or audiovestibular symptoms.
- •
Imaging is crucial in the evaluation of jugular foramen tumors and computed tomography (CT), MRI, and angiography provide complementary information in the diagnosis and preoperative planning for jugular foramen tumors.
- •
Surgical resection is often the primary treatment of nonparaganglioma jugular foramen tumors; however, the role of adjuvant radiotherapy and primary stereotactic radiotherapy is developing and promising.
- •
Stereotactic radiotherapy is an important adjuvant when subtotal resection is necessary or in cases of tumor recurrence.
| CN | Cranial nerve |
| CPA | Cerebellopontine angle |
| CT | Computed tomography |
| ELST | Endolymphatic sac tumor |
| HL | Hearing loss |
| JF | Jugular foramen |
| VHL | von Hippel-Lindau |
Epidemiology
Jugular paragangliomas are the most common tumors of the jugular foramen (JF) with an incidence of 1 per 1.3 million people per year (250 cases in the United States per year). Paragangliomas are not, however, the only tumors found in the JF. Schwannomas, meningiomas, and other tumors also grow in this location, although each is rare. As of 2005, there were only an estimated 200 cases of JF schwannoma reported in the English literature, and less than 100 cases of JF meningioma reported as of 2007. Even less common tumors that may involve or extend into the JF include endolymphatic sac tumors (ELST), chordomas, and chondrocytomas, and metastatic disease spread to the JF.
JF schwannomas are rare, with a reported incidence of 2.9% to 4% of all intracranial schwannomas and less than 1% of all temporal bone lesions. There are a limited number of small case series and reviews in the literature because of the rarity of these tumors. These tumors are reported to have an increased incidence in women and have an average age of incidence of 37 years. These tumors rarely present before the second or after the seventh decades of life. Presentation in the first or second decade of life should raise clinical suspicion for neurofibromatosis type 2.
JF meningiomas are very rare tumors that represent approximately 0.7% to 4% of all posterior fossa meningiomas. They are the third most common JF tumor following paraganglioma and schwannoma. Meningiomas can originate in the JF or may secondarily involve the JF with expansive growth, such as with some large petroclival meningiomas. Posterior fossa meningiomas are a rare group of tumors themselves, representing approximately 9% to 10% of all intracranial meningiomas. In a series of 161 posterior fossa meningiomas, only seven cases were identified as JF meningiomas. There is a strong female predominance and they usually occur in the fourth or fifth decade of life.
ELSTs are rare, low-grade adenocarcinomas of the endolymphatic sac that may extend through the temporal bone to the JF because of their infiltrative growth pattern. Although most ELSTs occur sporadically, they are a characteristic tumor of von Hippel-Lindau (VHL) disease with an incidence in VHL disease of 11% to 16%. VHL disease is an autosomal-dominant tumor syndrome with an incidence of approximately 1 in 36,000 births. The usual age at onset on VHL-associated ELST is in the third decade of life. ELSTs associated with VHL are bilateral in approximately 30% of cases. Sporadic ELST is usually diagnosed at a later age than VHL-associated tumors, and is usually unilateral.
Chordomas and chondrosarcomas are distinct entities but often grouped together because of their similar presentations, radiographic appearance, and anatomic location. These are slow growing, but locally aggressive, and rarely metastasize. Chondrosarcomas are cartilaginous tumors that make up approximately 11% of all bone tumors. Although these tumors are most commonly found in long bones, they involve the head and neck in approximately 10% of cases. They have been reported in a wide range of ages but tend to favor younger patients with no significant gender predilection. When chondrosarcomas involve the skull base they are most likely to arise at the petroclival junction and rarely involve the JF. According to a 2008 review by Sanna and colleagues, only 11 cases of chondrosarcoma arising from the JF were reported in the worldwide literature.
Cerebral metastasis occurs in approximately 15% to 35% of patients with cancer. In a series of 212 patients with primary nondisseminated malignant neoplasms, 22.2% of the patients were identified to have histopathologic evidence of temporal bone metastasis. Involvement of the skull base has been estimated to occur in approximately 4% of cases of systemic malignancy. Adenocarcinomas that are metastatic to the JF usually originate from the prostate, breast, kidney, or lung.
Epidemiology
Jugular paragangliomas are the most common tumors of the jugular foramen (JF) with an incidence of 1 per 1.3 million people per year (250 cases in the United States per year). Paragangliomas are not, however, the only tumors found in the JF. Schwannomas, meningiomas, and other tumors also grow in this location, although each is rare. As of 2005, there were only an estimated 200 cases of JF schwannoma reported in the English literature, and less than 100 cases of JF meningioma reported as of 2007. Even less common tumors that may involve or extend into the JF include endolymphatic sac tumors (ELST), chordomas, and chondrocytomas, and metastatic disease spread to the JF.
JF schwannomas are rare, with a reported incidence of 2.9% to 4% of all intracranial schwannomas and less than 1% of all temporal bone lesions. There are a limited number of small case series and reviews in the literature because of the rarity of these tumors. These tumors are reported to have an increased incidence in women and have an average age of incidence of 37 years. These tumors rarely present before the second or after the seventh decades of life. Presentation in the first or second decade of life should raise clinical suspicion for neurofibromatosis type 2.
JF meningiomas are very rare tumors that represent approximately 0.7% to 4% of all posterior fossa meningiomas. They are the third most common JF tumor following paraganglioma and schwannoma. Meningiomas can originate in the JF or may secondarily involve the JF with expansive growth, such as with some large petroclival meningiomas. Posterior fossa meningiomas are a rare group of tumors themselves, representing approximately 9% to 10% of all intracranial meningiomas. In a series of 161 posterior fossa meningiomas, only seven cases were identified as JF meningiomas. There is a strong female predominance and they usually occur in the fourth or fifth decade of life.
ELSTs are rare, low-grade adenocarcinomas of the endolymphatic sac that may extend through the temporal bone to the JF because of their infiltrative growth pattern. Although most ELSTs occur sporadically, they are a characteristic tumor of von Hippel-Lindau (VHL) disease with an incidence in VHL disease of 11% to 16%. VHL disease is an autosomal-dominant tumor syndrome with an incidence of approximately 1 in 36,000 births. The usual age at onset on VHL-associated ELST is in the third decade of life. ELSTs associated with VHL are bilateral in approximately 30% of cases. Sporadic ELST is usually diagnosed at a later age than VHL-associated tumors, and is usually unilateral.
Chordomas and chondrosarcomas are distinct entities but often grouped together because of their similar presentations, radiographic appearance, and anatomic location. These are slow growing, but locally aggressive, and rarely metastasize. Chondrosarcomas are cartilaginous tumors that make up approximately 11% of all bone tumors. Although these tumors are most commonly found in long bones, they involve the head and neck in approximately 10% of cases. They have been reported in a wide range of ages but tend to favor younger patients with no significant gender predilection. When chondrosarcomas involve the skull base they are most likely to arise at the petroclival junction and rarely involve the JF. According to a 2008 review by Sanna and colleagues, only 11 cases of chondrosarcoma arising from the JF were reported in the worldwide literature.
Cerebral metastasis occurs in approximately 15% to 35% of patients with cancer. In a series of 212 patients with primary nondisseminated malignant neoplasms, 22.2% of the patients were identified to have histopathologic evidence of temporal bone metastasis. Involvement of the skull base has been estimated to occur in approximately 4% of cases of systemic malignancy. Adenocarcinomas that are metastatic to the JF usually originate from the prostate, breast, kidney, or lung.
Presentation
All JF lesions have similar presenting signs and symptoms, which may include neuropathies of the cranial nerves (CNs) V-XII, especially the lower CNs IX-XI and audiovestibular dysfunction. A retrotympanic mass may be seen on otoscopy. Although glomus jugulare tumors are the most common type of JF tumor to present with this constellation of signs and symptoms, JF schwannomas and meningiomas often present similarly.
There are several different JF syndromes that are described based on differential involvement of the lower CNs. Although these syndromes are described as JF syndromes, lesions that cause these symptoms may occur anywhere along the course of the involved nerves from the brainstem to the extracranial site of innervation, not exclusively during their course through the JF. The constellation of lower CN deficits seen in JF syndromes have also been described for etiologies other than JF tumors including brainstem cerebral vascular accident and leptomeningeal processes (including tuberculosis, syphilis, and neurosarcoidosis). Abscess, trauma, and aneurysm may also result in a similar pattern of lower CN deficits.
- •
JF (Vernet) syndrome: Unilateral involvement of CN IX-XI resulting in loss of taste from the posterior one-third of the tongue; ipsilateral anesthesia of the palate, pharynx, and larynx; and ipsilateral weakness of vocal cords, palate, trapezius, and sternocleidomastoid.
- •
Posterior lacerocondylar (Collet-Sicard) syndrome: Involvement of IX-XI as in JF syndrome with addition of hypoglossal nerve (CN XII) involvement causing tongue weakness.
- •
Posterior retropharyngeal (Villaret) syndrome: Involvement of IX-XII as in posterior lacerocondylar syndrome but with the addition of sympathetic chain involvement leading to Horner syndrome.
JF meningiomas often present with hearing loss (HL), tinnitus, and a middle ear mass. Lower CN dysfunction is often present, although single nerve involvement seems to be more common than multiple nerve/JF syndromes. Other presenting signs and symptoms may include a mass in the neck or hoarseness. The presence of conductive HL is often caused by tumor extending into the middle ear and surrounding the ossicles without destruction or erosion, which is characteristic of tegmen tympani and JF meningiomas.
The presentation of JF schwannomas is variable depending on where the bulk of the tumor is located. The tumor may be predominantly intracranial, extracranial, or dumbbell-shaped in both compartments. The most common presentations involve HL, which is reported in up to 60% to 75% of patients, and lower CN deficits, reported in more than 50% of cases. However, if most of the tumor is intracranial with only mild involvement of the JF, the patient may present with minimal or no lower CN deficit and have primarily symptoms associated with intracranial mass including HL, tinnitus, and vertigo. A middle ear mass is rarely seen with these tumors. These tumors can also cause displacement of the facial nerve similar to vestibular schwannomas, and postoperative facial weakness has been reported in 20% to 25% of patients in some series.
The presentation of ELST is nonspecific and may be mistaken for other conditions. Chordomas and chondrosarcomas may present with headaches and diplopia, and trigeminal (CN V) neuropathy may also be present. Lower CN dysfunction is less common than with JF schwannomas and meningiomas.
Metastatic disease behaves more aggressively than JF tumors of benign histology and is more likely to cause cranial neuropathy. Metastatic disease is the most common cause of the JF syndrome. There is usually a rapid onset of symptoms as compared with the more insidious development of symptoms seen with JF paragangliomas, schwannomas, or meningiomas.
Evaluation and diagnosis
Although all JF lesions may have similarities in their clinical presentation, it is important to accurately diagnose the type of lesion. The behavior of the different tumor types is different and there are differences in preoperative evaluation, expected outcomes, and risk of recurrence. Imaging is the cornerstone of the diagnostic process.
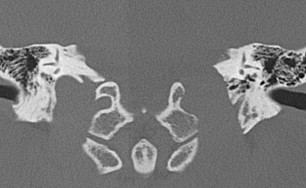
Preoperative imaging should include computed tomography (CT) and MRI, which provide complementary data for evaluating lesions of the JF. CT is particularly useful for evaluation of the osseous margins and identification of important bony landmarks for surgical planning ( Fig. 1 ). CT imaging should be performed with axial thin sections of 1.0 mm or thinner, without contrast, in bone algorithm (maximum edge enhancement), and include coronal reconstruction.
MRI allows for a more detailed evaluation of the tumor and the brain, posterior fossa, and soft tissues of the neck. MRI provides important information about the extension of the JF mass and the potential for evaluating vascular involvement. Evaluation of the skull base with MRI should be performed with axial and coronal thin section (4 mm or thinner) with T1- and T2-weighted sequences, thin section gadolinium-enhanced T1-weighted sequences with fat saturation, and diffusion-weighted sequences. The apparent diffusion coefficient series included with diffusion (diffusion weighted imaging [DWI] or diffusion tensor imaging [DTI]) imaging can be very helpful as a marker of tumor cellularity. Fat saturation is a useful technique for evaluating bone marrow involvement, particularly for tumors that extend into the petrous apex and erode the clivus. Postcontrast thin sections are helpful for tumor spread patterns, such as with perineural invasion, and foramen involvement.
For vascular lesions of the JF, such as paragangliomas and ELSTs, angiography is also useful. Angiography allows for assessment of the vascularity of the tumor and its relationship to the carotid artery. In the case of carotid artery encasement or occlusion, a balloon test can then be used to predict how well the patient would tolerate ligation of the carotid if necessary. The venous phase of angiography should also be evaluated to determine if there is intraluminal extension of the tumor mass or occlusion of the jugular bulb, jugular vein, or the sigmoid sinus. Moreover, venous angiography can be used to embolize the inferior petrosal sinus, which can minimize blood loss during resection because this sinus is otherwise difficult to manage until the jugular bulb has been opened.
Imaging characteristics
Meningiomas are solid, well-circumscribed, extra-axial masses. These tumors are usually sessile with a broad dural base because the intracranial spread along the dura is “en plaque” rather than globular in most cases. On CT imaging, there is commonly an increase in the density (sclerotic changes) of the bone adjacent to the meningioma ( Fig. 2 ). On MRI, there is usually intense, uniform enhancement, with an enhancing dural tail along the margin, and in comparison with glomus jugulare, an absence of flow voids ( Fig. 3 ). Meningiomas may arise within the JF, or they may originate in the posterior fossa and extend into the JF, termed primary or secondary JF meningiomas, respectively, based on their site of origin. Primary JF meningiomas have a unique growth pattern. They tend to be more infiltrative and often demonstrate a centrifugal growth pattern with expansion of the bony margins of the foramen along with local intraosseous extension. Secondary JF meningiomas have a growth pattern that is more typical of other intracranial meningiomas, with growth characterized by the soft tissue mass component with minimal bone involvement.
Schwannomas typically show a smooth-corticated margin. Schwannomas cause expansion of the JF but do not invade the marrow space. This type of JF expansion is well-visualized on CT. On MRI, schwannomas typically show isointensity on T1, hyperintensity on T2 sequences, and there are no flow voids (distinguishing from glomus jugulare) or dural tails (distinguishing from meningioma). Occasionally, cystic degeneration (intratumoral cysts) may be seen within the schwannoma. Schwannomas avidly enhance on MRI T1 gadolinium-enhanced images. JF schwannomas are typically lobulated, well circumscribed, and elongated following the course of the CNs ( Fig. 2 ). These tumors follow the course of CNs IX-XI, as they arise from these nerves, and therefore extend along the nerves superomedially into the posterior fossa toward the lateral brainstem, or inferiorly toward the nasopharyngeal carotid space. Up to 90% of these tumors are reported to arise from the glossopharyngeal or vagus nerve. A case series of 10 JF schwannomas revealed involvement of these locations with no involvement of the temporal bone or clivus. This constrained spread along the CNs distinguishes JF schwannomas from the infiltration of JF meningiomas and glomus jugulare tumors into the surrounding skull base. Minimal or no tumor blush is seen in these tumors on angiography, which would not typically be indicated if a schwannoma is suspected.
Tumor extension for schwannomas is described by the Kaye and Pellet classification. Kaye and colleagues described three classes of JF schwannoma: (1) intracranial with minimal extension into bone (type A), (2) primarily within bone regardless of intracranial component (type B), and (3) primarily extracranial with minimal extension into bone or into the posterior fossa (type C). Pellet and colleagues proposed the addition of a fourth class of JF schwannoma describing dumbbell-shaped tumors with extracranial and intracranial extensions linked via the JF (type D). The type and extent of tumor extension is important to identify on preoperative imaging to identify the optimal surgical approach to the JF that maximizes tumor resection and neural preservation.
Characteristic findings of chordomas or chondrosarcomas that extend to the JF include irregular bony erosion and occasional calcifications within the mass seen on CT, and hypointense or isointense in signal on T1-weighted and hyperintense on T2-weighted MRI. Gadolinium enhancement may be heterogeneous or homogeneous and the tumor may have a “soap bubble” appearance. These pathologies cause expansile changes to the skull base, whereas approximately 50% of chondrosarcomas demonstrate a chondroid matrix within the lesion. Chordomas and chondrosarcomas have a similar radiographic appearance, but the location may distinguish these tumors. Chordomas of the skull base arise in the midline in the clivus, because they originate from notochord remnants. Chondrosarcomas have a more laterally located center and grow toward the midline because they arise from mesenchymal cells or rests of cartilage within the skull base. The value in distinguishing these tumor types relates to differences in prognosis and the role of adjunct radiotherapy.
ELSTs are often confused for other JF lesions based solely on their imaging characteristics that are similar to other infiltrative tumors, such as paragangliomas. These tumors are centered in the endolymphatic sac at the posterior petrous surface, however, and demonstrate a destructive pattern of growth best seen on CT, with central calcification, and a posterior rim of calcification. MRI characteristics of ELSTs typically include hyperintensity on T1-weighted images and heterogeneous enhancement with gadolinium, similar to glomus jugulare, with a “salt and pepper” appearance ( Fig. 4 ) from hypervascularity. There is heterogenous hyperintensity on T2-weighted images (see Fig. 5 ).



