Meningioma
If the reader were to think of just one tumor type that would cross multiple specialty lines it would probably be the meningioma. The meningioma is a very important tumor in the fields of ophthalmology, neuro-ophthalmology, neurosurgery, and head and neck surgery because of its frequency, its destructive effect on vision, the difficulties associated with its surgical eradication, its persistent course, and its sometimes fatal termination.
A major event in the study of this tumor, since the third edition of this text was published, has been the availability of neuroimaging in all major hospital/medical centers for the demonstration of this tumor. This has made possible the visualization of the soft-tissue component of the tumor that was previously unseen by standard orbital radiography. This provided earlier diagnosis of meningioma when compared to imaging methods used before. Imaging of the tumor has been further refined by the use of magnetic resonance and the subsequent clinical use of the contrast agent gadoliniumdiethylene triamine pentacetic acid. The latter further increased the diagnostic yield. Surgeons kept pace with these advances in diagnosis by providing more extensive surgical exposures and using microsurgical techniques for the removal of the tumor, particularly those meningiomas along the skull base. More recently steroid receptors have been identified on the meningioma cell that may have implications for hormone therapy of these tumors. Finally, the use of the Gamma Knife (stereotactic radiosurgery) offers another therapeutic option for the management of selected meningiomas.
As has been the practice in the previous editions of this text, we are chiefly concerned with meningiomas that by reason of proptosis, imaging characteristics, or the findings uncovered at the time of surgery were known to have encroached upon the orbital cavity. This includes those tumors that seem to arise within the orbit (primary type) and those that extend into the orbit from an intracranial source (secondary type). The primary group may be subdivided into those tumors arising within the sheath of the intraorbital portion of the optic nerve and those arising elsewhere in the orbit, particularly along the orbital face of the greater wing of the sphenoid bone. In the latter, the position of the meningioma is almost exclusively intraorbital. The secondary tumors with orbital encroachment usually arise from the sphenoid ridge, the basofrontal region, the area around the sella, or extracranial sites such as the paranasal sinuses. In general, the source of any given meningioma can be judged by the tumor’s principal blood supply. In cases where the meningioma arises along a cranio-orbital junction, such as the optic canal or superior orbital fissure, the tumor is tabulated in the “secondary” group.
Incidence
The importance of meningioma in orbital oncology is underlined by the 178 cases with orbital involvement in our 50-year survey (Table 3.3). They constitute 9.9% of our total of 1,795 orbital tumors. Meningioma is the fourth most common primary orbital tumor and the third most common secondary orbital tumor (Table 3.3). According to the classification noted in the preceding text, 64 (36.0%) are primary in the orbit and 114 (64.0%) are the secondary type. Of the 64 of the primary type, 30 (47%) are of optic nerve sheath origin. The remaining of the primary type arise from the orbital surface of the sphenoid bone and encroach on the intraorbital space by a combination of hyperostosis of affected bone and expansion of soft-tissue tumor.
In bygone years, orbital meningioma was almost always considered a disease of adults. Younger patients who also presented with monocular visual loss followed by unilateral proptosis were assumed to have an optic nerve glioma, based on age selectivity of the two tumors. Over time an increasing number of cases of orbital meningioma have been recorded in children. When Walsh (1970) first reported a series of seven pediatric optic nerve sheath meningiomas, he commented that the tumors were “aggressive.” Karp et al. (1974), reflecting the Armed Forces of Pathology experience with 10 patients, believed these tumors were highly invasive but not malignant. Wright et al. (1989), commented that four of the six children studied already had intracranial extension at the time of surgical excision. The patients described and the period of this article predate high-resolution neuroimaging, and subtle degrees of intracranial extension were not detected
prior to surgery but the point remains that most had intracranial extension. The case of Cibis et al. (1985) is of particular interest. This was a 13-year-old girl with intraocular extension of an optic nerve sheath meningioma who had had visual symptoms since 2 years of age. Initially, the patient was assumed to have an optic nerve glioma. This patient also had neurofibromatosis type-22 (NF-2). The Mayo Clinic series included five patients (two girls, three boys) who were 18 years or less of age (range 5.8 to 18 years). In our patients, the histopathology was benign but described as invasive. A recurring theme amongst all the reports is the frequency of NF-2. Of Walsh’s 7 patients, at least 2 had NF-2, whereas 2 of Karp’s 10 patients, 1 of Wright’s 6 patients, and 3 of the 5 patients from Mayo had NF-2. One of the Mayo patients is shown in Figure 18.1. Some of the data in the older literature concerning childhood meningioma might be challenged, because meningeal hyperplasia secondary to an underlying optic nerve glioma was sometimes erroneously diagnosed as meningioma. Despite this disclaimer, it is apparent that orbital meningioma can arise in children and should be considered in the differential diagnosis of the more common optic nerve glioma. Also, the presence of an optic nerve sheath meningioma in a child should call to mind the possibility of an underlying NF-2.
prior to surgery but the point remains that most had intracranial extension. The case of Cibis et al. (1985) is of particular interest. This was a 13-year-old girl with intraocular extension of an optic nerve sheath meningioma who had had visual symptoms since 2 years of age. Initially, the patient was assumed to have an optic nerve glioma. This patient also had neurofibromatosis type-22 (NF-2). The Mayo Clinic series included five patients (two girls, three boys) who were 18 years or less of age (range 5.8 to 18 years). In our patients, the histopathology was benign but described as invasive. A recurring theme amongst all the reports is the frequency of NF-2. Of Walsh’s 7 patients, at least 2 had NF-2, whereas 2 of Karp’s 10 patients, 1 of Wright’s 6 patients, and 3 of the 5 patients from Mayo had NF-2. One of the Mayo patients is shown in Figure 18.1. Some of the data in the older literature concerning childhood meningioma might be challenged, because meningeal hyperplasia secondary to an underlying optic nerve glioma was sometimes erroneously diagnosed as meningioma. Despite this disclaimer, it is apparent that orbital meningioma can arise in children and should be considered in the differential diagnosis of the more common optic nerve glioma. Also, the presence of an optic nerve sheath meningioma in a child should call to mind the possibility of an underlying NF-2.
Secondary orbital meningiomas, specifically the sphenoid wing meningioma, can also occur in the pediatric population. We have seen four patients, all boys with this meningioma. The age range was 5.5 to 15.6 years. All were of meningothelial type and one invaded the bone and the others invaded orbital fat. One patient had neurofibromatosis type-1 (NF-1) and another had NF-2. Right lower lid swelling was noticed in a 5-year-old boy in 1985. Biopsy through anterior orbitotomy revealed meningothelial meningioma. A more definitive craniotomy was done 2 months later. From 1986 through 1991 he had three recurrences, all treated by craniotomy. When we examined him in 1992, he had no light perception in the right eye, had a frozen, enophthalmic globe, and a palpable mass along the lateral inferior orbital rim. He was treated with another craniotomy, an exenteration, a rectus abdominus free flap and 5,000 cGy external beam radiation therapy. To date (2004) he has not had a recurrence.
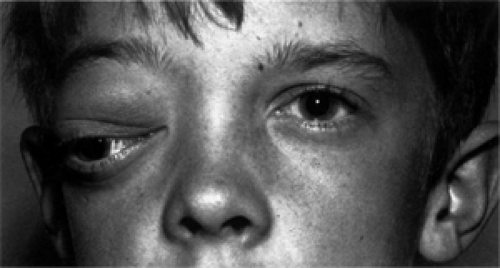 Figure 18.1 Recurrent meningioma: A 13-year-old boy with proptosis of right eye commencing at age 10 years. First craniotomy for removal of intraorbital meningioma was at age 12. Two additional craniotomies and one orbitotomy were performed over the next 2 years. At age 21 recurrent orbital tumor and proptosis of right eye gave the patient a gargoyled appearance. However, visual acuity in the affected eye was still 20/40. No further follow-up until age 35 when patient was seen elsewhere. Clinical diagnosis at this time was bilateral hearing loss secondary to acoustic neurinomas. The visual acuity was not measured. Computed tomography scan showed probable meningioma of right orbit, lesser wing of sphenoid bone, and frontal convexity, left cavernous sinus, and anterior falx. |
The prevalence of meningiomas in women is well known. In the Mayo Clinic series, 72% (129/178) are women, a female-to-male ratio approximating 3:1. Five of the 33 men are clustered in the first and second age decades.
The tumor’s prevalence in women suggests some hormonal association. Indeed, several recent studies document the presence of receptors for estrogen, progesterone, and somatostatin in meningioma cells (Halper et al., 1989; Cahill et al., 1984; Tilzer et al., 1982; Huisman et al., 1991; Arena et al., 2004). Specific receptors for progesterone were found in 76% of 70 meningiomas studied by Lesch and Fahlbusch (1986). This is consistent with the observed progression of meningiomas during pregnancy and breast-feeding. Such cases associated with pregnancy are frequently reported in the literature. Wan et al. (1990) reviewed this subject and reported three patients whose symptoms were exacerbated during pregnancy. Estrogen receptors are found less consistently and tend to be present in men.
Schrell et al. (1990) have a different view. They examined 50 human cerebral meningiomas and concluded that estrogenic and progestrogenic targets are of negligible significance. They inferred that female sex steroids are not primarily involved in the proliferative rate of these tumors and are of limited value as markers for adjuvant medical therapy.
The most recent work with RU-486 (mifepristone) is currently unpublished data (Gruenberg et al.). Their data on 18 patients suggests that there is no clinical effect in 33%, slows down progression in another 33%, and leads to a slight decrease in tumor volume with visual field improvement in the remaining 33% (Sadun A., unpublished data, personal communication, 2004).
The discrepancy amongst the reports of both estrogen and progesterone receptors may be related to differences in laboratory techniques. Nevertheless, meningiomas can be clearly conceived as targets of hormonal action on their receptor types, but the hormonal effects have still to be defined in detail (Schrell, 1992). Schrell also concluded that meningioma is, indeed, a tumor of endocrinologic interest but is not an endocrine tumor.
Somatostatin receptors have been detected in virtually all human meningiomas (Koper et al., 1992; Huisman et al., 1991). The effects of hormonal manipulation have been controversial and currently there are no therapeutic applications related to this receptor (Cavalla and Schiffer, 2001; Arena et al., 2004).
At one time the age distribution of orbital meningiomas was thought to be bimodal, with one age peak in the first two decades and the other peak in the 40 to 50 age range (Karp et al., 1974; Eggers et al., 1976). With the advent of computed tomography (CT) scan and magnetic resonance imaging (MRI) it is now apparent that orbital meningiomas may occur at any age. Table 3.4 shows a broad age distribution at the time of surgical diagnosis of both primary and secondary meningiomas. Eighty-seven percent (154/178) of cases were concentrated in the fourth through seventh decade. The age range of the 64 primary meningiomas was 6 months to 83 years with a median age of 40 years. The age range of the 114 secondary meningiomas was 4 months to 75 years with a median age of 52 years. The slightly greater prevalence of primary meningiomas in the younger age range is consistent with other surveys of meningioma in the literature.
In the total 178 patients in our study core, the laterality ratio is 78:56, right to left orbit. Bilateral orbital meningiomas were present in three patients at the time of presentation. In several additional patients the disease became bilateral during the course of their long survival.
NF was recorded in three patients with primary orbital meningiomas (NF-2) and two patients with secondary orbital meningiomas (one NF-1 and one NF-2).
In the 8-year period (1980–1987) since the third edition of this text, 505 intracranial meningiomas without orbital invasion were surgically verified at the Mayo Clinic. In this same interval of time, 41 meningiomas with orbital involvement were identified. The ratio of intracranial meningiomas to orbital meningiomas in the 8-year interval, therefore, is 505:41 (roughly 12:1). The grand total of all meningiomas is the sum of these two groups or 546 cases. The incidence of primary orbital meningiomas (nine cases) relative to total meningiomas then becomes 1.6%. The incidence of orbital meningiomas (both primary and secondary types) relative to all meningiomas is 7.5%.
We have not attempted a specific comparison of incidence data of the present Mayo Clinic series, which consists of consecutive cases with pathologic verification, with other surveys of meningioma in the literature. Most other series of meningiomas differ from ours in size, the data in some is based on a combined sample of proved and assumed cases of meningioma, others include only selected clinical cases, and in still others the data is based on cases selected from a file of pathologic specimens. Such comparisons, which were attempted in the previous two editions, give inconsistent conclusions, not unlike a comparison of oranges and apples.
Clinical Features
The presenting symptoms and signs associated with meningiomas, either originating in or encroaching on the orbit, are numerous and diverse; they include the gamut of proptosis, a decrease of visual acuity of 20/40 or less, pallor of the optic disk, detection or awareness of a visual field defect, passive edema of the eyelid, disturbance of ocular motility, papilledema, headaches or orbital pain, a visible or palpable mass, opticociliary (chorioretinal)shunt vessels, paralytic blepharoptosis, and seizures, roughly in the order of their frequency. Other symptoms or signs, such as chemosis and afferent pupillary defects, are secondary to the size of the orbital mass or the degree of visual loss, respectively. Most patients usually have a minimum of three of the above signs or symptoms at the time of presentation. However, none of these signs or symptoms is specific for meningioma. The degree or frequency of any of these clinical features depends on the duration of the meningioma and its location.
Although unilateral proptosis is the most frequent presenting sign, it is neither present nor is it always the initial clinical feature in all cases. Some degree of visual impairment often preceded proptosis in the anamnesis of the patient. The amount of proptosis was greater in primary orbital meningiomas than with meningiomas secondarily encroaching on the orbit.
Impairment of visual acuity (20/40 or less) paralleled proptosis in its diverse degree. Some patients noted only a visual blur of one eye that could only be defined by demonstrating a depression of the visual field by perimetry. A few patients described visual obscurations that were gaze evoked, indicating a large mass in the orbital apex that was compressing the optic nerve. Of more serious note were patients whose vision in the affected eye was reduced to light perception, light projection, or no light perception at the time of presentation. The prevalence of such a serious visual loss was slightly greater in the secondary orbital meningiomas when compared to primary orbital meningiomas.
Pallor of the optic disk of the affected eye, the fourth most common clinical feature, was equally prevalent in the primary and secondary orbital meningiomas. Its presence at the time of the patient’s presentation indicates that the meningioma either had been present longer or was greater in extent than the patient suspected.
Visual field loss is another clinical feature that varies widely in extent. From an initial, overall depression, the visual field may progress through a dense arcuate defect, a central scotoma associated with an irregular peripheral contraction, a bizarre irregular altitudinal defect, to a complete loss of central field associated with a few peripheral islands of remaining vision. Such a visual field loss in the affected eye, when associated with a hyperostosis of the sphenoid bone, was a positive indication for an exploratory surgical procedure in the years before the present neuroradiologic imaging modes.
Boggy edema of the eyelids, usually more marked in the lower eyelid, is a peculiar manifestation of some long-standing meningiomas and occurs more often with this lesion than any other orbital tumor of similar duration
or size. The affected lid is pale and soft, and the swelling resembles the puffiness of the eyelids seen with myxedema in women of the same age-group (see Fig. 18.2). It occurs in both the primary and secondary types of orbital meningioma. It is puzzling why edema of the eyelids occurs in some orbital meningiomas but not in other meningiomas of equal size or location. The cause of the edema is not known, although it is commonly assumed to be secondary to a stasis of venous flow through the exit channels of the orbit. In some cases, intravascular clumps of meningioma may be responsible for the edema (see Fig. 18.3).
or size. The affected lid is pale and soft, and the swelling resembles the puffiness of the eyelids seen with myxedema in women of the same age-group (see Fig. 18.2). It occurs in both the primary and secondary types of orbital meningioma. It is puzzling why edema of the eyelids occurs in some orbital meningiomas but not in other meningiomas of equal size or location. The cause of the edema is not known, although it is commonly assumed to be secondary to a stasis of venous flow through the exit channels of the orbit. In some cases, intravascular clumps of meningioma may be responsible for the edema (see Fig. 18.3).
 Figure 18.2 Intraorbital meningioma: A 49-year-old woman with marked edema of left eyelid and proptosis of approximately 6 years’ duration. Vision of left eye reduced to counting fingers. |
A derangement of motility of the affected eye may be secondary to the mechanical effect of a bulky orbital mass, to an infiltration of the extraocular muscle by a meningioma that has escaped from the dural sheath of the intraorbital optic nerve, or to an entrapment of the III, IV, or VI cranial nerves by a tumor invading the superior orbital fissure or the area of the cavernous sinus. As a rule the subjective or objective importance of the motility disturbance is overshadowed by other clinical manifestations of meningioma at the time of presentation.
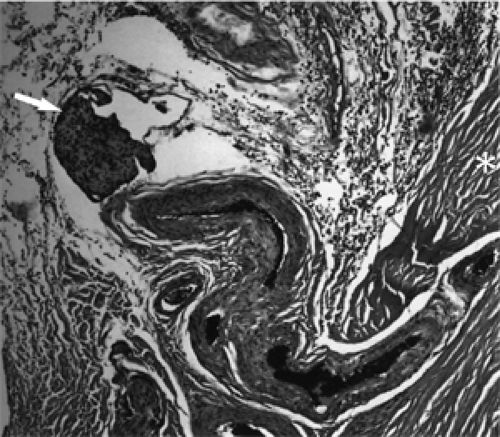 Figure 18.3 Meningioma: Clump of tumor within vessel wall (arrow). Asterisk designates sclera (× 100). |
The presence of papilledema at the time of the patient’s presentation is of interest, because it occurs much more frequently in primary orbital meningioma when compared to a percent prevalency in the secondary type of meningioma.
Fortunately, orbital pain or nagging headaches are an infrequent manifestation of meningioma at the time of presentation. However, when present, it is of major concern to the patient and is the major reason for the patient’s seeking consultation. This clinical manifestation does not seem to have any specific localizing significance.
The meningiomas with which we are concerned are, with few exceptions, residents of the posterior orbit. Seldom will the mass be palpable in the anterior portion of the orbit unless the tumor has reached considerable size. However, in a minority of cases the mass may be present in the temporal fossa. Here the mass is smooth, not tender, and may give the area of the temple a “fullness” or domed appearance. These masses are the result of a slow lateral expansion of a meningioma of the greater wing of the sphenoid bone. A mass in the temporal fossa is very suggestive of a meningioma because there is no other orbital tumor that so frequently presents in this manner in an adult.
A type of presentation not mentioned in the preceding discussion is the intraocular meningioma. This tumor represents the less common, anterior extension of a meningioma of the sheath of the intraorbital optic nerve. This route of extension is very infrequent, and when it occurs, the case frequently is reported in the literature. Such was the situation in one case of the Mayo Clinic series that was reported by Henderson and Campbell (1977).
Briefly, this case was a 70-year-old woman whose sightless, anesthetic, proptosed eye was enucleated in 1976. The retrobulbar space was filled with a poorly circumscribed cellular meningothelial meningioma. Apart from a very pale optic disk, the posterior pole of the enucleated eye appeared normal. However, microscopy revealed focal meningiomatous invasion of the sclera and a tumor nodule within the choroid. Seventeen additional cases of intraglobal extension of a meningioma were also referenced in the publication. Earlier we cited a subsequent case of intraocular extension of a meningioma in a 13-year-old girl (Cibis et al., 1985).
Bilaterality was noted in five female patients with secondary meningiomas who first presented at Mayo Clinic for evaluation. Their ages were 18, 62, 65, 68, and 76 years. All patients had bilateral pallor of the optic disks. All had experienced slowly progressive visual loss. Several of the elderly patients had experienced visual loss over a period of several
decades, but the cause of the visual loss was not detected by standard roentgenography. Diagnosis finally was made by the CT scan of the 1970s. The intracranial origins of the tumor were usually the surface of the lesser wing of the sphenoid bone. One tumor originated from the falx, and one tumor arose from the greater wing of the sphenoid bone.
decades, but the cause of the visual loss was not detected by standard roentgenography. Diagnosis finally was made by the CT scan of the 1970s. The intracranial origins of the tumor were usually the surface of the lesser wing of the sphenoid bone. One tumor originated from the falx, and one tumor arose from the greater wing of the sphenoid bone.
A few additional remarks are appropriate concerning the clinical aspects of those patients with an exacerbation of meningioma during pregnancy. Initially, in the first few months of pregnancy, such patients will note some unilateral blurring of vision associated with headache. A diagnosis of unilateral retrobulbar optic neuritis might be considered at this stage. However, this innocuous onset quickly gives way to a rapid decline in vision in the affected eye with pallor of the optic disk. This leads to a discovery by neuroimaging of an enhancing mass somewhere along the course of the optic nerve, with or without hyperostosis of the bone, which is suggestive of meningioma. Usually, an effort is made to manage these patients conservatively until the pregnancy is successfully completed. Intervention, either surgical or with radiation therapy is deferred unless the loss of vision is very rapid and severe.
In some patients the symptoms may improve and the meningioma stabilizes after delivery. Surgical resection of the presumed meningioma may then be postponed. In such cases the meningioma may recur with increased severity during a subsequent pregnancy.
In terms of counseling a patient with a meningioma regarding pregnancy, management issues are not clear in the literature. Wan et al. (1990) did mention that management of pregnant patients with meningioma should be tailored to the individual, and we would agree with this. In general, although we do not discourage pregnancy or breast-feeding, patients should be apprised of the potential risks.
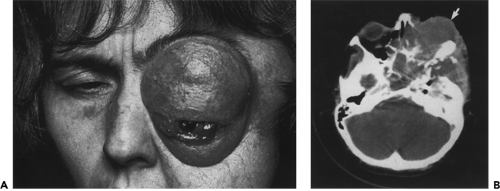 Figure 18.4 A: Recurrent meningioma. A 48-year-old woman who had had two previous resections of a sphenoid wing meningioma 12 and 6 years prior to this photo. A left lateral tarsorrhaphy had been performed at the age of 45 to protect the bulging, displaced eye. The left eye was blind. The tumor involves the face below the left eyebrow, and there is also a bulging mass in the left temple. The left orbit was exenterated in the course of a third surgical resection of the tumor. B: Axial computed tomography scan 10 years later shows recurrent tumor (arrow) involving left orbit, paranasal sinuses, middle, and anterior intracranial fossae, temporalis fossa, and parasellar region.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|