Key points
- •
Children with conductive hearing loss (CHL) are identified at several possible diagnostic points of care: newborn hearing screening, parental concern for hearing loss, hearing screening in the pediatrician’s office, otoscopic examination either in the pediatrician’s or in the otolaryngologist’s office, or school hearing screening examination.
- •
The evaluation of any child with hearing loss involves a thorough history with careful otoscopic examination to include pneumatic otoscopy.
- •
Audiological assessment is a critical part of the evaluation and depends on the child’s age and cooperability (see article by Singleton and Waltzman elsewhere in the issue).
- •
Any child with an unexplained CHL (normal ear canal, tympanic membrane, middle ear space) should undergo radiologic evaluation, usually high-resolution computed tomography (HRCT) of the temporal bone at some point (see later discussion and article by DeMarcantonio and Choo elsewhere in the issue).
- •
Management of the child with acquired CHL depends on the cause and may range from simple cerumen disimpaction to ventilation tube insertion, to more complex chronic ear surgery for tympanic membrane perforation or cholesteatoma.
- •
Management of the child with congenital CHL may include observation with monitoring of the hearing, an individual education (Individual Education Plan [IEP]) or 504(c) plan, amplification (conventional or bone conducting), or surgery. Management depends greatly on the diagnosis, degree of hearing loss, relevant anatomy, parental decision making, and educational and psychosocial factors.
Introduction
While sensorineural hearing loss (SNHL) is far more common in adults, CHL accounts for 90% to 95% of all childhood hearing loss, with middle ear effusion (MEE)/otitis media with effusion (OME) far outpacing all other causes. Whether OME causes lasting deficits in speech and language development remains unclear, probably due to the transient, fluctuating nature of the associated hearing loss, involvement of one or both ears, mild degree of associated hearing loss, and medical and surgical options for management. Fixed, moderate, or moderate to severe congenital CHL such as that caused by congenital aural atresia (CAA) or other ossicular abnormality (eg, congenital stapes ankylosis) is rarer but may cause lasting deficits in speech and language development and educational progress, especially if bilateral and not evaluated and managed early and properly.
Prevalence estimates of congenital SNHL range in the 1 to 3 per 1000 range, whereas estimates for congenital CHL are less well reported; however, clearly, this type of hearing loss, caused by some obstruction, dysfunction, or maldevelopment of the ear canal, eardrum, or middle ear impedance system, is also relatively rare. In a study of 234 Australian infants referred for diagnostic testing from a newborn hearing screening program, prevalence of CHL in the newborns was 2.97 per 1000 while the prevalence of middle ear pathology (with or without CHL) was 4.36 per 1000. As one investigator noted, “In the literature pertaining to CHL in children, the emphasis is on cause rather than severity, making prevalence data difficult to compare.” The ongoing and lasting effects of both congenital and acquired CHL in children, especially with regard to OME, have been studied exhaustively, yet no clear conclusions have been made.
Options for hearing habilitation in children with congenital CHL include observation with monitoring—both hearing and academic progress—and possible individualized education plan (IEP)/504(c); amplification, either conventional or through bone conduction technology; and surgery. In children with bilateral moderate or moderate to severe fixed CHL such as that seen in CAA, amplification and/or surgical intervention is strongly recommended to support normal speech and language development, but controversy remains on the ideal management of the child with unilateral CHL.
This article provides the clinician with guidelines to inform the evaluation and management of childhood CHL—acquired and congenital as well as unilateral and bilateral.
Introduction
While sensorineural hearing loss (SNHL) is far more common in adults, CHL accounts for 90% to 95% of all childhood hearing loss, with middle ear effusion (MEE)/otitis media with effusion (OME) far outpacing all other causes. Whether OME causes lasting deficits in speech and language development remains unclear, probably due to the transient, fluctuating nature of the associated hearing loss, involvement of one or both ears, mild degree of associated hearing loss, and medical and surgical options for management. Fixed, moderate, or moderate to severe congenital CHL such as that caused by congenital aural atresia (CAA) or other ossicular abnormality (eg, congenital stapes ankylosis) is rarer but may cause lasting deficits in speech and language development and educational progress, especially if bilateral and not evaluated and managed early and properly.
Prevalence estimates of congenital SNHL range in the 1 to 3 per 1000 range, whereas estimates for congenital CHL are less well reported; however, clearly, this type of hearing loss, caused by some obstruction, dysfunction, or maldevelopment of the ear canal, eardrum, or middle ear impedance system, is also relatively rare. In a study of 234 Australian infants referred for diagnostic testing from a newborn hearing screening program, prevalence of CHL in the newborns was 2.97 per 1000 while the prevalence of middle ear pathology (with or without CHL) was 4.36 per 1000. As one investigator noted, “In the literature pertaining to CHL in children, the emphasis is on cause rather than severity, making prevalence data difficult to compare.” The ongoing and lasting effects of both congenital and acquired CHL in children, especially with regard to OME, have been studied exhaustively, yet no clear conclusions have been made.
Options for hearing habilitation in children with congenital CHL include observation with monitoring—both hearing and academic progress—and possible individualized education plan (IEP)/504(c); amplification, either conventional or through bone conduction technology; and surgery. In children with bilateral moderate or moderate to severe fixed CHL such as that seen in CAA, amplification and/or surgical intervention is strongly recommended to support normal speech and language development, but controversy remains on the ideal management of the child with unilateral CHL.
This article provides the clinician with guidelines to inform the evaluation and management of childhood CHL—acquired and congenital as well as unilateral and bilateral.
Acquired conductive hearing loss
Otitis Media with Effusion
Prevalence
By far the most common cause of acquired CHL in children, OME is defined as the presence of fluid in the middle ear without the signs or symptoms of acute, active infection. It is estimated that at any given point in time, approximately 20% of young children have a MEE, with nearly all children having at least 1 episode during their childhood. OME commonly follows an upper respiratory tract infection or is a sequela of acute otitis media and is usually self-limited. Certain populations have a higher prevalence of OME than the general population of young children. Children with Down syndrome have poor eustachian tube function because of decreased motor tone, predisposing them to persistent OME. OME also occurs with increased prevalence in children with cleft palate. In fact, it is nearly universal in children with cleft palate because of abnormal insertion of the levator veli palatini and tensor veli palatini muscles on the eustachian tube, resulting in poor active opening. Other risk factors include male gender, immunodeficiency, ciliary dyskinesia, bottle feeding, cigarette smoke in the house, increased number of siblings in the house, and perhaps the most important—day care.
Almost 85% to 100% of ears affected by OME have a type B, or flat, tympanogram. A type B tympanogram can occur without the presence of MEE, as in the case of tympanosclerosis, or after tympanoplasty. Pneumatic otoscopy should be the primary means by which OME is diagnosed.
Effect of otitis media with effusion on speech and language development
OME causes a CHL by limiting mobility of the tympanic membrane and ossicles, which results in decreased sound transmission through the middle ear. The impact of OME on hearing can range from no hearing loss (0 dB HL) to moderate range hearing loss (55 dB HL). The average hearing loss associated with OME has been estimated at 28 dB HL, but in approximately 20% of children, it exceeds 35 dB HL. Even mild degrees of SNHL have been shown in children to cause speech and language delay, so one could presume that a CHL secondary to chronic MEE could similarly impact a child’s speech and language development. Hearing loss may be suggested by seeming lack of attentiveness, behavioral changes, excessive volumes on television or listening devices, or failure to respond to normal conversational volumes. It has been previously demonstrated that parents cannot reliably detect mild degrees of hearing loss in children.
If OME has persisted in both ears for 3 months or longer (chronic OME), there is a lower chance of spontaneous resolution. In a meta-analysis, Rosenfeld and Kay found that only 26% of patients with chronic OME showed resolution by 6 months with a 33% resolution rate by 1 year.
Despite evidence of hearing loss associated with OME, there is no evidence that untreated children with OME fair worse developmentally, specifically with respect to language, than do children treated with early tympanostomy tube insertion. No studies have been performed on children with an established speech, language, or developmental delay comparing outcomes with versus without surgical treatment of OME.
Management of otitis media with effusion—American Academy of Otolaryngology guidelines
The decision to place ventilation tubes depends on multiple factors including the child’s speech and language development to date, the presence of craniofacial abnormalities, recurrence of infections, and the presence of other risk factors cited earlier. The American Academy of Otolaryngology Clinical Practice Guidelines on OME and Tympanostomy Tubes in Children were published in 2004 and 2013, respectively (selected, summarized recommendations are given in Box 1 ).
- 1.
Clinicians should distinguish the child with OME who is at risk for speech, language, or learning problems from other children with OME and should more promptly evaluate hearing, speech, language, and need for intervention. This procedure includes children with previously documented delay and those with craniofacial abnormalities, as described above.
- 2.
Clinicians should manage the child with OME who is not at risk with watchful waiting for 3 months from the date of effusion onset (if known) or from the date of diagnosis (if onset is unknown). This fact is supported by the evidence that in most cases OME is self-limited.
- 3.
Antihistamines and decongestants are ineffective for OME and are not recommended for treatment. Antimicrobials and corticosteroids do not have long-term efficacy and are not recommended for routine management. Devices that promote autoinsufflation, however, are useful in children who are old enough to participate.
- 4.
Hearing testing is recommended when OME persists for 3 months or longer, or at any time that language delay, learning problems, or a significant hearing loss is suspected in a child with OME. Language testing should be conducted for children with hearing loss.
- 5.
Children with persistent OME who are not at risk should be reexamined at 3- to 6-month intervals until the effusion is no longer present, significant hearing loss is identified, or structural abnormalities of the eardrum or middle ear are suspected. If a child has a moderate hearing loss, greater than 40 dB HL, then treatment is recommended. If the hearing loss is mild (20–35 dB HL) or normal (0–20 dB HL), then repeat testing can be performed in 3 to 6 months.
- 6.
When a child becomes a surgical candidate, tympanostomy tube insertion is the preferred initial procedure; adenoidectomy should not be performed unless a distinct indication exists (nasal obstruction, chronic adenoiditis). Repeat surgery consists of adenoidectomy plus myringotomy, with or without tube insertion. Tonsillectomy alone or myringotomy alone should not be used to treat OME. Tympanostomy tube insertion has been shown to improve hearing by an average of 6 dB at 6 months. Adenoidectomy has a demonstrated benefit as adjuvant treatment in children requiring reinsertion of tympanostomy tubes, with a 37% to 47% reduction in OME and a 50% reduction in need for reoperation.
Conductive Hearing Loss Associated with Chronic Otitis Media
Chronic otitis media (COM) is defined as any ear with a tympanic membrane perforation—either with or without cholesteatoma. This section provides guidelines for the management of chronic ear disease in children; it is not meant as an exhaustive review, rather a framework on which to base decisions to optimize outcomes for children with this relatively common disease.
Tympanic membrane perforation
In children, tympanic membrane perforation is most often the result of a retained ventilation tube or a sequela of ventilation tube insertion. Perforations are typically anterior-inferior, the site of ventilation tube placement, and as such, do not cause significant CHL. CHL associated with tympanic membrane perforation is generally in the low frequencies. Larger perforations are associated with larger CHLs, although the location of perforation has not been shown to affect the degree of CHL.
Before surgical repair, children should undergo audiometric evaluation with air and bone conduction thresholds to evaluate the degree of CHL. Success of pediatric tympanoplasty depends on both intrinsic (patient-derived) and extrinsic (surgeon-derived) factors. Intrinsic factors that should be optimized include patient age, presence of otorrhea, ongoing eustachian tube dysfunction, status of the contralateral ear, health of the child, and size and location of the perforation; extrinsic factors include surgical approach, technique, and surgeon experience. Perhaps the greatest predictor of success in pediatric tympanoplasty is age; children younger than 4 years have worse outcomes than older children, but the optimal age for successful tympanoplasty remains uncertain. The status of the contralateral ear is another important consideration.
Techniques for pediatric tympanoplasty are wide ranging and include fat graft myringoplasty (with or without hyaluronic acid ), underlay, and overlay/lateral grafting techniques. The key to successful tympanoplasty involves evaluating extrinsic and intrinsic factors, optimizing those factors, and practicing practice-based learning—examining personal outcomes and making changes based on a surgeon’s own outcomes.
A particular technique that has been successful for closure of one perforation may not carry the same success rates for other perforations. In general, small, posterior central perforations can be managed with a myringoplasty (eg, fat plug) or underlay technique. Marginal perforations do better with underlay or lateral surface grafting. Large, total, or subtotal perforations are generally managed with a lateral surface technique. Other perforations can be managed with any of the techniques, depending on surgeon comfort and experience, and on optimizing those intrinsic factors that play into the success of tympanoplasty in children.
Tympanosclerosis
Tympanosclerosis is the result of recurrent or chronic middle ear inflammation resulting in fibrosis, which can lead to fixation of the ossicles and associated CHL. Tympanosclerosis can be seen innocuously in the tympanic membrane (myringosclerosis) and not associated with CHL, or it can be an insidious cause of progressive CHL as it immobilizes the ossicular chain. Any child with a history of recurrent ear infections and/or myringosclerosis with progressive CHL should be suspected of having ossicular hypomobility due to tympanosclerosis.
Management may include observation with monitoring of the hearing, amplification (conventional or bone conducting), or surgical exploration. Although tympanosclerosis generally has multiple loci in the middle ear, hearing results of lateral chain fixation secondary to tympanosclerosis, in which the surgeon used a partial or total ossicular replacement prosthesis, trend better than those of tympanosclerotic stapes fixation, in which mobilization or partial/total stapedectomy has been performed. HRCT of temporal bone can be helpful in elucidating the site of ossicular fixation and can give the surgeon and family preoperative and prognostic information on chances of hearing rehabilitation with surgery. Stapedectomy, either partial or total, can be successful in restoring hearing but is not advised in the presence of a tympanic membrane perforation—a staged approach must be undertaken to close the perforation before opening the footplate.
Of course, any operation to remove or address fibrosis or scar tissue risks further fibrosis, so long-term follow-up is required.
Cholesteatoma
Cholesteatoma refers to stratified squamous epithelium (skin) growing in the middle ear and may be congenital or acquired. Cholesteatoma causes CHL either by mass effect on the ossicular chain or by bony erosion of the ossicles. Primary acquired cholesteatoma starts as a tympanic membrane retraction, potentially as a consequence of eustachian tube dysfunction, either in the posterior pars tensa (sinus cholesteatoma, Fig. 1 ) or in the pars flaccida (attic cholesteatoma, Fig. 2 ). The retracted area of the eardrum forms a pocket lined by healthy stratified squamous epithelium that fills with desquamated epithelium (keratin), and the pocket expands as it fills with keratin. Cholesteatoma may also develop from an eardrum perforation as the skin migrates into the middle ear rather than closing the perforation (secondary acquired cholesteatoma).
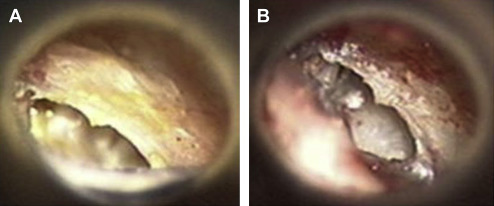
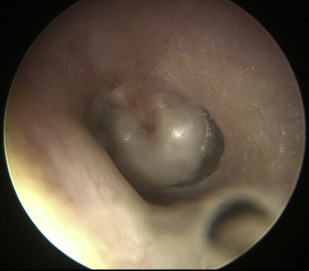
Congenital cholesteatoma is usually seen as a pearly mass behind the anterosuperior quadrant of an intact tympanic membrane, thought to originate from an epithelial rest in the middle ear ( Fig. 3 ). Congenital cholesteatoma may also arise from the mastoid and reach a large size before being diagnosed. Congenital cholesteatomas represent about 4% of all pediatric cholesteatomas. Cholesteatomas are erosive, and acquired cholesteatoma often presents with otorrhea and chronic, recalcitrant infection. Congenital cholesteatoma presents with a CHL and a white retrotympanic mass.
Comprehensive management of pediatric cholesteatoma, often more invasive and more aggressive than in the adult, is surgical and beyond the scope of this article (see Weber, Sismanis and colleagues, and Brackmann and colleagues ). The primary goal of surgery is a clean, safe, dry ear with no recurrent or residual disease. Recurrence rates are higher in children than in adults possibly related to continued immaturity of the eustachian tube. Secondary goal is rehabilitation of the associated CHL and may involve ossiculoplasty (either at the time of cholesteatoma resection or as a staged procedure), conventional amplification, osseointegrated bone conducting implant, or even preferential seating in class with or without a frequency modulation (FM) system.
Effect of chronic otitis media on hearing and speech/language development
The above-mentioned entities generally affect school-aged children and as a result, are not thought to have a dramatic impact on speech and language development during the critical early years of life. The degree of CHL associated with cholesteatoma, tympanic membrane perforation, and tympanosclerosis varies with the extent of disease. An average pure tone air conduction of 41.3 dB has been reported in a review of 158 cholesteatoma ears. As noted, CHL varies with size but not location of tympanic membrane perforation. Cholesteatoma and tympanosclerosis typically affect hearing in only 1 ear, and having a normal contralateral ear, children may cope well with this degree of hearing loss. Careful monitoring of academic progress, preferential seating in class, consideration of an FM system, and amplification are strategies used to rehabilitate children with chronic ear disease. Unilateral CHL in the severe range has been shown to affect academic performance, but the CHL associated with cholesteatoma may not be severe, unless there is complete ossicular discontinuity.
The priority in treating cholesteatoma is to achieve a safe, dry ear; good hearing results are often achievable with ossicular chain reconstruction. Conventional hearing aids and bone conducting hearing aids are also appropriate and successful in hearing rehabilitation.
Congenital conductive hearing loss
Overview
Congenital anomalies of the middle ear are rare causes of CHL in children and are classically divided into major (CAA, defined by absence or stenosis of the external auditory canal [EAC] and some middle ear ossicular underdevelopment) and minor malformations. Minor malformations are characterized by a normal auricle, intact and patent EAC, intact tympanic membrane, and a deformation, fixation, or disruption of ossicular conduction. The hearing loss associated with these minor malformations, including congenital stapes ankylosis, persistent stapedial artery, malleus bar/malleus fixation, and absent oval window can range from mild to severe, can be missed on newborn hearing screening, and may not be diagnosed until the child can sit for behavioral testing. Some of these anomalies include those that are also associated with CAA, such as malleus bar and malleoincudal fixation.
Development and Embryology of the Middle Ear
The eustachian tube develops during the third week of gestation from the endoderm of the first branchial pouch, with the lateral end of the pouch forming the middle ear cleft. Pneumatization of the tympanic cleft begins around week 10. The tympanic membrane forms during the 28th week and is derived from all 3 layers of primitive tissue: ectoderm forming the outer squamous layer, mesoderm forming the middle fibrous layer, and endoderm forming the medial mucosal layer.
The malleus and incus are a single mass at 6 weeks, separating at 8 weeks. The head and neck of the malleus, as well as the body and short process of the incus, are derivatives of Meckel’s cartilage (first branchial arch). The manubrium of the malleus, long process of the incus, and the superstructure of the stapes are derivatives of Reichert’s cartilage (second branchial arch). Arrest in development of the first branchial arch underlies CAA, which is accompanied by a fused malleus-incus complex, absent manubrium, and often normal stapes bone (second arch derivative), as well as mandibular hypoplasia (hemifacal microsomia).
The stapes begins to form at 4 to 5 weeks from the blastema. Cranial nerve VII divides the blastema into the stapes, interhayle (future stapedius tendon), and laterohayle (future pyramidal eminence). The tympanic portion of the footplate develops from the second branchial arch cartilage, and the vestibular portion of the footplate (lamina stapedalis and annular ligament) originates from the otic capsule. The 2 develop independently and eventually fuse. The interhayle develops into the stapedial muscle and tendon, and the laterohayle becomes the posterior tympanic wall. Ossification begins at week 19 and proceeds from both the stapes superstructure and the otic capsule and, under normal conditions, does not involve the vestibular portion of the footplate, which includes the annular ligament, which should remain cartilaginous throughout life.
An important concept to understand in the embryology of the ear is that the ear canal and middle ear develop from the branchial apparatus; the cochlea, vestibule, labyrinth, and central auditory nervous system structures develop from the otocyst, from the neuropore. The implication is that most patients with congenital CHL have just that—a conductive and not a mixed or sensorineural loss. That said, patients with CAA can have morphologic inner ear abnormalities identified on computed tomographic (CT) scan, but these abnormalities do not necessarily translate into an SNHL. Proper audiometric evaluation with air and bone conduction testing will best characterize the hearing.
Patient Evaluation
Diagnosing CHL in the child with grade II or III microtia and associated CAA ( Fig. 4 ) is very straightforward—the disability is easily seen on physical examination and appropriate testing can be performed. After newborn hearing screening, recommended testing protocols include otoacoustic emissions testing in the normal ear or the ear with a minor malformation, air and bone conduction auditory brainstem response (ABR) testing in the normal ear, and bone conducting testing in the atretic ear if unilateral, and bone conducting ABR if bilateral. The child with a CHL secondary to a minor malformation or the child with a normal auricle and CAA can be deceiving, but the hearing loss is usually identified at newborn hearing screening, or occasionally later in life, at a school screening. Careful binocular microscopic examination either in the office or under anesthesia with air and bone conduction ABR testing can diagnose CAA or minor malformation and CHL.



