Purpose
To evaluate the potential benefit and risk of proton beam therapy in the treatment of symptomatic retinal papillary capillary hemangioma.
Design
Retrospective interventional case series.
Methods
This study included patients presenting with symptomatic exudative retinal papillary capillary hemangioma with or without association with von Hippel–Lindau disease. All patients were treated either as a first or a secondary treatment option by proton beam therapy between 2001 and 2009. The minimum follow-up was 30 months.
Results
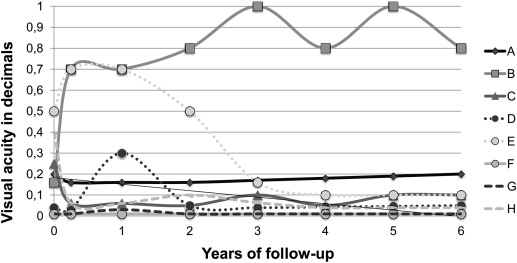
Eight eyes of 8 patients (3 male and 5 female, with a mean age of 36 years [range 22–80 years]) were treated for symptomatic papillary retinal hemangioma. The median interval between onset of macular edema and proton beam therapy was 1.7 months (range 0.5–3.3 months). The median follow-up period was 84 months (range 32–106 months) between proton beam treatment and last follow-up. Exudation completely resolved in all but 1 patient after 4.2 months on average (range 2.8–7.2 months). Mean visual acuity prior to proton beam irradiation was 0.7 logMAR (0.2 DIN (DIN 58220 norm)) (range 2-0.3 logMAR) and declined to 0.8 logMAR (0.16 DIN; range 2-0.1 logMAR) at last follow-up examination (no statistical significance, P = .071).
Conclusion
The anatomic outcome after proton beam therapy for retinal papillary hemangioma is convincing, whereas functional outcome may be compromised because of tumor location, long-persisting macular edema, extensive exudation, and poor initial visual acuity. In patients with extended retinal detachment surgical intervention was still necessary. Although proton beam therapy is proven to be a therapeutic option, treatment will remain challenging.
Retinal capillary hemangiomas are also known as retinal hemangioblastomas. These lesions must be distinguished from choroidal hemangiomas, which may be associated with Sturge-Weber syndrome. Retinal capillary hemangiomas can occur in isolation or as part of von Hippel–Lindau (VHL) disease. As retinal capillary hemangiomas are often the earliest and most frequent disease manifestations seen in VHL, accurate diagnosis by the ophthalmologist plays an important role in the initiation of appropriate screening and treatment.
VHL disease is an autosomal-dominant disorder with variable penetrance and expression that is caused by mutations of the VHL gene, a tumor suppressor gene located at the short arm of chromosome 3. Aside from having retinal capillary hemangiomas, individuals with VHL disease also have a high incidence of renal cell carcinoma, central nervous system hemangiomas, pheochromocytomas, and other tumors. Renal cell carcinoma is the leading cause of mortality in patients with VHL disease. Even though retinal capillary hemangiomas are classified as benign, significant visual morbidity may be caused through either exudative or tractional effects on the surrounding retina. Although many therapeutic modalities exist, treatment of symptomatic cases can be challenging, especially in juxtapapillary retinal hemangiomas. These vascular hamartomas grow on the optic nerve head or within the juxtapapillary area. Three growth types have been classified: the endophytic, exophytic, and sessile forms. The differential diagnosis can sometimes be difficult, resulting in misdiagnoses such as papilledema, papillitis, granulomatous disease, or subretinal neovascularization. Thus, after careful ophthalmoscopy, fluorescein angiography is the next essential step hinting at correct diagnosis of juxtapapillary/papillary hemangioma. A very early hyperfluorescence of the feeding vessels in the arterial phase and a staining in the late phase, sometimes combined with a delicate to distinct leakage, can be observed. Treatment modalities for juxtapapillary capillary hemangioma include laser photocoagulation, photodynamic therapy, and brachytherapy, leading to such complications as arcuate scotoma, optic ischemia and retinal capillary occlusion compromising the functional benefit, and radiation-induced optic neuropathy, respectively. External beam radiotherapy showed promising results, but only in a very limited number of patients. Until now, treatment of juxtapapillary capillary hemangioma has remained a therapeutic dilemma.
This study reports long-term results after proton beam therapy in papillary capillary hemangioma to evaluate the potential benefit and long-term risks of this treatment modality.
Methods
This retrospective interventional case series included patients with visual loss, metamorphopsia, photopsia, or scotoma attributable to symptomatic retinal papillary hemangioma with or without association with VHL disease. The ethics committee of the Charité-Universitätsmedizin Berlin waived the need for Institutional Review Board approval owing to the retrospective design of the study. It was ensured that the study and data accumulation were in conformity with all country, federal, or state laws; informed consent was obtained; and the study was in adherence to the tenets of the Declaration of Helsinki.
All patients were treated between January 2001 and December 2009 in the Department of Ophthalmology, Charité University Medicine Berlin, Campus Benjamin Franklin (patient recruitment, preparation, and follow-up) and the Helmholtz-Zentrum Berlin. The minimum follow-up was 30 months.
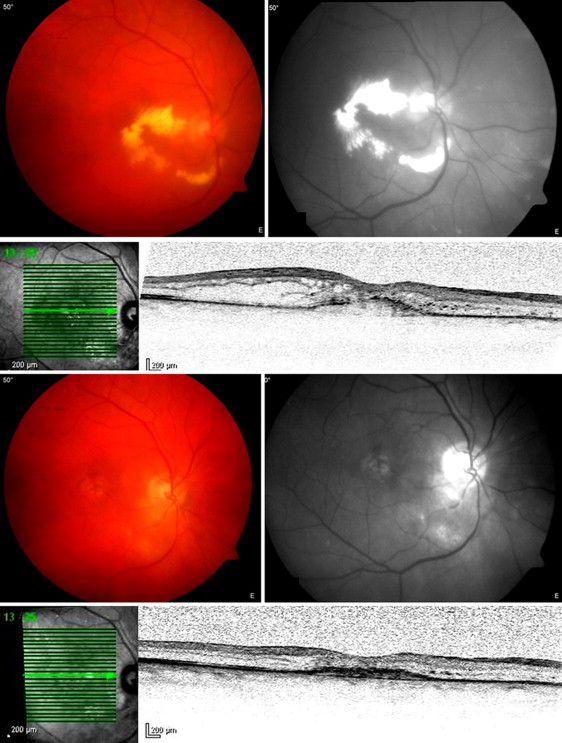
The retinal papillary capillary hemangioma was diagnosed by slit-lamp examination, ophthalmoscopy, and fluorescein angiography. All patients presenting with retinal capillary hemangiomas were tested for VHL gene mutations. In patients presenting positive test results a screening for VHL-associated tumors located elsewhere was started. Based on medical records, imaging data, surgical reports, and schemes of irradiation, the investigated endpoints of this study were the functional and anatomic outcome. Visual acuity is described as decimals and was measured as logMAR. In agreement with the MARAN protocol, light perception was added as 2.1 logMAR, hand motion as 2.0 logMAR, and counting fingers as 1.9 logMAR. Tumor height and tumor diameter were measured by B-scan ultrasonography.
Local complications attributable to radiotherapy radiation optic neuropathy, radiation retinopathy, neovascular glaucoma, enucleation rate, and further surgery were recorded.
Proton Beam Therapy
The setting of the proton beam therapy used in retinal papillary capillary hemangiomas was similar to the established treatment for choroidal melanomas. Because of lack of established data regarding the dose of proton beam treatment for papillary capillary hemangioma, the dose was adapted to the dose described by Palmer and Gragoudas for retinal capillary hemangioma (2800–4000 cGy divided in 4 to 5 fractions) and the dose used for circumscribed choroidal hemangioma (on average 18 Gy) by Zografos and associates. Accordingly, the dose for proton beam therapy in our study was, in total, 20 CGE (cobalt gray equivalent; 1 CGE = 1.1 Gy, taking a radiobiological effectiveness of 1.1 into account), given in 4 fractions of 5 CGE on 4 sequential days. Prior to irradiation, 2.5-mm-diameter tantalum clips were sutured to the sclera to demarcate the margins of the tumor. Three-dimensional modeling of the tumor (clinical target volume), based on localization measurements, by ultrasound, funduscopy, and computed tomography scans; treatment planning; and dose calculation were performed with the treatment planning systems EYEPLAN version 1.2 and (since 2006) OCTOPUS version 4.4 (PRECISIS AG, Heidelberg, Germany). During treatment the head was immobilized by a combination of an individually molded face mask and a bite block to ensure the correct position. The patient had to focus on an external light source. In case of movement, the therapy could immediately be intermitted.
Statistics
Statistical analysis was performed with SPSS 20.0 (IBM, New York, USA). A Fisher exact test was used to detect differences in this small number of patients.
Results
Eight consecutive patients with visual loss attributable to solitary retinal papillary hemangioma were included in the study. There were no peripheral lesions identified. There were 3 male and 5 female patients. The mean age was 36 years (range 22–80 years). The median follow-up period was 84 months (range 32–106 months) between proton beam treatment and last follow-up.
Two patients showed a systemic association to VHL disease.
Tumor Characteristics
Before proton beam therapy median tumor height was 2.0 mm (range 1.3–4.7 mm), which decreased to 1.26 mm at time of last follow-up (1.0–2.2 mm: P < .001). Initial median largest tumor diameter was 6.25 mm (range 4.5–8.8 mm). All hemangiomas grew from the optic nerve head and reached toward the fovea with a median tumor–fovea distance of 1.25 mm (0–4.1 mm). One papillary hemangioma was located nasally while the remaining 7 were located on the temporal side of the optic nerve head ( Table ).
| Patient | Sex | Age at Diagnosis | Growth Type | Initial Tumor Height (mm) | Initial Tumor Diameter (mm) | Tumor–Fovea Distance (mm) | Tumor–Disc Distance (mm) | VHL Disease |
|---|---|---|---|---|---|---|---|---|
| 1 | M | 57 | Exophytic | 1.4 | 6.1 | 0 | 0 | No |
| 2 | F | 26 | Exophytic | 1.4 | 6.4 | 4.1 | 0 | No |
| 3 | F | 27 | Exophytic | 2 | 8.8 | 0 | 0 | No |
| 4 | F | 28 | Endophytic | 2 | 7.5 | 0 | 0 | No |
| 5 | M | 22 | Endophytic | 1.5 | 5 | 1.1 | 0 | Yes |
| 6 | M | 44 | Endophytic | 4.7 | 7.2 | 2.5 | 0 | Yes |
| 7 | F | 45 | Endophytic | 3.4 | 4.7 | 1.7 | 0 | No |
| 8 | F | 80 | Exophytic | 1.3 | 4.5 | 1.4 | 0 | No |
Anatomic Features
At the initial inclusion into the study all patients presented with macular edema. Furthermore, 3 out of 8 patients presented with an exudative retinal detachment. The retinal detachment affected the fovea in all cases; 2 quadrants were detached in 1 patient. A total exudative retinal detachment was detected in 2 patients. These patients required at least 1 vitrectomy after proton beam therapy to reattach the retina.
Exudation completely resolved in all but 1 patient after 4.2 months on average (range 2.8–7.2 months). This 1 patient received a single injection of bevacizumab because of persisting macular edema; however, the outcome was unsuccessful. Macular edema was stable over 3 years subsequently while the patient remained under observation.
Radiation Therapy/Surgical Treatment Before Proton Beam Therapy
The total dose delivered to the tumor and to the optic disc was 20 CGE in all patients. The median interval between onset of macular edema and proton beam was 1.7 months (range 0.5–3.3 months). Proton beam therapy was the first-line treatment in 6 patients and the secondary treatment in 2 patients. In the primary treatment group median time between onset of macular edema and proton beam therapy was 1.6 months (range 0.5–3.3 months). One of the 2 patients receiving proton beam as a secondary therapy initially presented with an exudative retinal detachment comprising 2 quadrants. Vitrectomy, including endolaser photocoagulation, triamcinolone acetonide, penetrating diathermy, and gas tamponade, was performed. In the second patient presenting with a total exudative retinal detachment laser photocoagulation was performed 3 times, following intravitreal bevacizumab and triamcinolone acetonide. In both patients treatment did not succeed, so proton beam therapy was indicated. Time interval between onset of macular edema and proton beam therapy was 1.5 months in the first patient and 3.0 months in the second patient ( Figure 1 ).
Functional Results
Mean visual acuity prior to proton beam irradiation was 0.7 logMAR (0.2 DIN) (range 2–0.3 logMAR) and declined to 0.8 logMAR (0.16 DIN; range 2–0.1 logMAR) at last follow-up examination (no statistical significance, P = .071). There was no correlation between the functional outcome and radiation-induced complications, either to optic neuropathy or to retinopathy. An initial visual acuity lower than 1.3 logMAR (0.05 DIN) remained stable in all patients (n = 3). Mean visual acuity dropped in all patients presenting with tumors located on the temporal side of the optic disc (n = 7) from 0.7 logMAR (0.2 DIN; range 2–0.3 logMAR) before proton beam therapy to 1.1 logMAR (0.08 DIN; range 2–0.7 logMAR) at final examination ( P = .095). One patient presenting with a tumor located on the nasal side of the optic nerve head improved from 0.8 logMAR (0.16 DIN) initially to 0.1 logMAR (0.8 DIN) at final follow-up examination ( Figure 2 ).