Purpose
To report delayed-onset peripheral ulcerative keratitis (PUK) following alkali injury.
Design
Retrospective case series.
Methods
setting : Single institution (Cornea and External Disease Service, Moorfields Eye Hospital). participants : Six eyes of 5 patients with PUK and associated anterior scleritis that had a history of ocular alkali injury. observation procedure : Patients were identified among PUK patients seen at Moorfields Eye Hospital over a 20-year period. main outcomes measures : Patients’ demographics, clinical features, treatment, and outcomes.
Results
Recurrent PUK with scleritis following alkali burns occurred in 5 male patients/6 eyes (median age: 22 years, range 18–38) several years after the chemical trauma (average: 6.4 years; range 3–12). Management of PUK in these patients was similar to PUK arising from other etiologies.
Conclusions
In this series of patients there was no evidence of an underlying vasculitic cause for the PUK. A localized autoimmune response may, however, be involved in the pathogenesis of these cases, as seen in an animal model of chemical injury or in late mustard gas keratitis. We hope that this case series will bring this newly described condition to the attention of ophthalmologists and that this may assist in their treatment, which, in this series, required systemic immunosuppressive therapy.
Peripheral ulcerative keratitis (PUK) is a destructive inflammatory process leading to corneal ulceration and stromal destruction, which involves primarily the juxtalimbal region. PUK is an uncommon disease, with a reported incidence of 3 cases per million per year. It is commonly present in association with conjunctival, episcleral, or scleral inflammation. The associated anterior scleritis can be diffuse, nodular, or necrotizing.
An underlying systemic condition, usually a vasculitis, is responsible for over half of the cases of PUK. The most common vasculitides associated with PUK include rheumatoid arthritis; the ANCA-positive vasculitides, such as granulomatosis with polyangiitis and microscopic polyangiitis; and systemic lupus erythematosus. PUK may also be infectious in etiology. Bacteria, fungi, herpes simplex virus, and Acanthamoeba have all been reported to cause PUK and scleritis by direct invasion or immune mechanisms. Exposure or mechanical trauma to the ocular surface can also cause PUK. Therefore PUK has multiple causes, and the underlying pathobiology may well differ between these.
The pathogenesis of PUK associated with vasculitis, although not thoroughly elucidated, relies on the abnormal activation of T cells with production of a cell-mediated and antibody response against corneal autoantigens.
In this retrospective case series, we report 5 cases of idiopathic PUK occurring several years after an alkali chemical burn.
Methods
This is a descriptive case series of patients with late-onset PUK following alkali ocular injury. Institutional review board and ethics committee approval was obtained for this study. Consecutive cases were selected from the PUK patients followed between 1992 and 2012 in Professor John Dart’s clinic, Cornea and External Disease Service at Moorfields Eye Hospital, London, United Kingdom. The medical records of 5 patients presenting with signs and symptom of delayed-onset PUK following ocular alkali injury were reviewed. A summary of the patients’ characteristics is reported in the Table .
| Characteristic | Case 1 | Case 2 | Case 3 | Case 4 | Case 5 |
|---|---|---|---|---|---|
| Sex | M | M | M | M | M |
| Age at trauma | 22 | 22 | 38 | 18 | 29 |
| Ocular comorbidities | None | None | Amblyopia | None | None |
| Systemic comorbidities | None | None | Crohn disease | Depression | None |
| Type of chemical injury | Ammonia | Ammonia | Ammonia | Ammonia | Ammonia |
| Cultures | Neg | Neg | Neg | Neg | Neg |
| Autoimmunity screening | Neg | Neg | Neg | Neg | Neg |
| Time from injury (y) | 4 | 3 | 8 | 5 | 12 |
| Involvement | Unilateral | Unilateral | Unilateral | Unilateral | Bilateral |
| Systemic treatment | Prednisolone, methylprednisolone (IV), cyclosporin, mycophenolate, infliximab | Prednisolone, cyclosporin, mycophenolate | Prednisolone, cyclosporin | Prednisolone | Prednisolone, cyclosporin, mycophenolate |
| Known episodes of inflammation | 3 | 2 | 2 | N/D | 2 |
| Average time between episodes of inflammation (y) | 0.4 | 2 | 2 | N/D | 2.5 |
| Surgical procedures | AMG (inlay and overlay) | LK (x3), DLC | Glue, PK, cataract extraction, upper lid skin graft | None | Cataract extraction |
| Resolution | Yes | Yes | Yes | N/D | Yes |
| Final CDVA | HM | HM | 20/40 | HM | 20/30 in both eyes |
None of the cases had microbiological evidence of infectious PUK or clinical and laboratory evidence of underlying autoimmune diseases, with the exception of 1 patient with a history of Crohn disease. No exposure, dry eye, or impaired corneal sensation was present in any of the eyes included in the study.
Summary of 2 representative cases is reported below. Description and images of Cases 1, 3, and 4 are reported as Supplemental Text and Supplemental Figure (available at AJO.com ).
Description of Representative Cases
Case 2
This subject suffered an alkali chemical injury (ammonia) to his right eye in 1986 at the age of 22. He had been originally seen in 1989 elsewhere for a recurrent sclerokeratitis and lipid deposition in the peripheral stroma. When he came to our attention in 1994, he had a large epithelialized descemetocele under a central tarsorrhaphy. As the corneal epithelial phenotype was found to be conjunctival according to the impression cytology cytokeratin profile, we carried out a successful lamellar keratoplasty using full-thickness tissue and including half of the donor limbus in the graft with diathermy of the major trunks of corneal neovascularization.
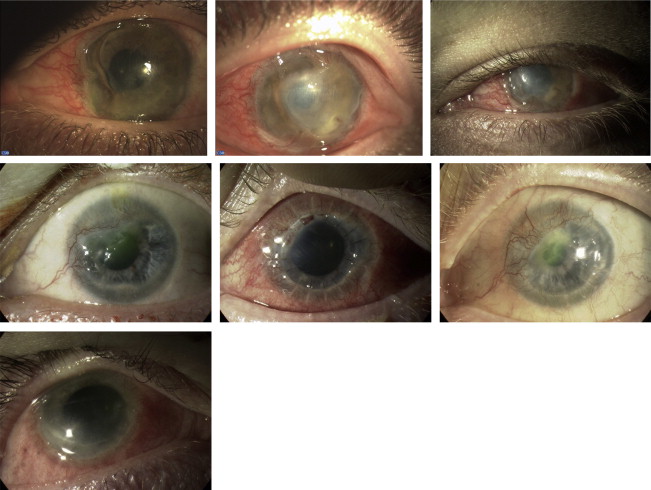
Unfortunately, the patient developed a recurrence of the sclerokeratitis 1 year later (1996) with lipid keratopathy and extensive stromal thinning, for which he was started on oral flurbiprofen and steroids ( Figure , Top left). Ultimately, a repeat lamellar keratoplasty was performed in June 1999, which completely epithelialized in a few weeks. In 2000 the patient began using a cosmetic contact lens, even though his visual acuity in the affected eye was 20/30 with the aid of a rigid gas-permeable contact lens.
In the following years the patient started suffering from persistent corneal epithelial defects, leading to corneal stromal melt and ultimately a necrotizing peripheral sclerokeratitis with a large superotemporal scleral rupture and uveal prolapse in 2007 ( Figure , Top center), for which a large tectonic sclerocorneal graft was necessary (January 2007). Control of the postoperative inflammation required the use of oral immunosuppression (prednisolone 50 mg daily, cyclosporin A 150 mg twice daily, and mycophenolate 1 g twice daily), which was tapered slowly over time and discontinued in early 2009. The eye ultimately stabilized with hand motion vision, residual bullous keratopathy, and several areas of scleral thinning ( Figure , Top right, detail). Over the years the patient also developed secondary glaucoma that required topical treatment as well as diode laser cyclophotocoagulation in October 2009.
Case 5
This 29-year-old patient suffered a chemical injury to both eyes with ammonia as a result of an assault in 1996. The patient had very few ocular symptoms until late 2008, when he started experiencing increasing redness, photophobia, and discomfort in both eyes unresponsive to topical steroids. He was referred to Moorfields Eye Hospital in July 2009. At his first appointment in our clinic the patient had 20/20 vision in both eyes with bilateral posterior blepharitis and diffuse scleral injection, particularly in the inferior quadrants; inferior corneal thinning with peripheral neovascularization; and an epithelial defect in his left eye. Investigations for autoimmune disease were negative. The patient was started on topical cyclosporin A 2% and systemic immunosuppression with oral prednisolone (80 mg daily) and oral cyclosporin A (250 mg twice daily). There was complete resolution on the oral immunosuppressive treatment, which was eventually discontinued in early 2009 ( Figure , Bottom left, Bottom right) when the patient developed hepatotoxicity. Topical treatment did not control the disease, which reactivated in November 2012. On this occasion he was given another course of oral steroids (prednisolone 40 mg daily) with oral mycophenolate (1 g twice daily). Oral cyclosporin A (150 mg twice daily) was later added to the treatment to achieve better control of the inflammation. Posterior subcapsular cataract extraction was ultimately performed in both eyes with a final visual acuity of 20/30 bilaterally.
Common Features
Common features of the cases described in this series are reported in the Table .
All chemical traumas were caused by alkali compounds containing ammonia and occurred in male young adults (median age: 22 years; range: 18–38). PUK occurred after an average of 6.4 years (range: 3–12 years) from the ammonia burn. No significant other ocular comorbidities were present and all patients were healthy, with the exception of 1 patient with Crohn disease. There was no evidence of an infectious or autoimmune etiology in these cases. The PUK had a relapsing-remitting course in all cases, with multiple episodes of inflammation occurring over the years. Both eyes were involved in only 1 case of bilateral chemical injury. The medical and surgical management of these cases was similar to PUK secondary to other causes. The final visual outcome in these cases was dismal, with the exception of Cases 3 and 5.
Discussion
In this case series, we present 5 cases of PUK with scleritis occurring long after an alkali (ammonia) chemical injury to the eye. This late complication of alkali injury occurred in young male subjects, was noninfectious, and was characterized by recurrent episodes of sclerokeratitis. In all cases, systemic immunosuppressive treatment was necessary to achieve long-term control of the inflammation.
Noninfectious keratitis with corneal ulceration can occur at different stages in chemical injuries, with variable etiology.
In the immediate phase (1–3 days), corneal ulceration can be caused by direct epithelial or stromal lysis and limbal ischemia with secondary stem cell deficiency. The modified Hughes classification of alkali injuries is based on the degree of stromal haze and limbal ischemia. With the exception of Case 1 (grade 4), the severity of the initial alkali injury could not be determined. However, all patients in this series had completely epithelialized after the initial alkali injury.
In the acute phase (1–3 weeks), keratitis is often secondary to the influx of leukocytes in the corneal stroma drawn by corneal neovascularization. In the late stages (>3 weeks), recurrent epithelial defects or frank ulcers are usually secondary to epithelial instability secondary to limbal stem cell deficiency or to neurotrophic/exposure damage to the ocular surface. In the cases reported in this series all ulcers occurred years after the chemical injury. None of the cases had evidence of infection, dry eye, or exposure keratopathy. Although we could assume that a variable degree of limbal stem cell deficiency was present in all cases, we do not believe that delayed-onset PUK, with associated scleritis, could have been attributable solely to partial limbal stem cell failure, as this has not been previously reported in this context.
The etiology of PUK is most frequently infectious or autoimmune. None of the patients in this series exhibited clinical or microbiological evidence of infection. There was no evidence of any autoimmune conditions associated with PUK, with the exception of Case 3 with Crohn disease, which has been associated with PUK. In this patient, we cannot exclude that the keratitis could have been secondary to inflammatory bowel disease rather than to the previous alkali injury. The patient was older than the other cases but shared a similar long interval between the chemical injury and the onset of PUK. PUK in Crohn disease usually accompanies reactivations of the intestinal inflammation; in our patient the keratitis occurred when the bowel disease was quiescent, which was the situation throughout the course of the ocular disease.
Two cases of PUK have been previously reported after mechanical ocular trauma. A case of peripheral ulcerative keratitis with necrotizing scleritis was described 1 month after surgical repair for a traumatic scleral rupture. The patient had hepatitis C and mixed cryoglobulinemia. The surgical procedure, rather than the traumatic event, seemed to be linked to the postoperative scleritis. A further case of PUK caused by a retained hard contact lens in the superior fornix was reported by Bhatt and associates. Repeated mechanical trauma or local action of bacterial endotoxins were the supposed triggers of PUK in this patient.
The immunopathogenesis of vasculitic PUK has been thoroughly investigated but not yet clarified fully. Local deposition of immune complexes, a cell-mediated and humoral autoimmune response to corneal antigens, and dysregulation of T suppressor lymphocytes have all been advocated in the initiation of the autoimmune inflammatory process. Neoangiogenesis and lymphangiogenesis occurring in the peripheral cornea provide the afferent and efferent arm for the autoimmune response, with priming of naïve lymphocytes, migration to lymphoid organs, and then homing of targeted lymphocytes to the peripheral cornea.
A similar autoimmune mechanism could be hypothesized in these cases of post–alkali injury PUK. Ammonium hydroxide, the toxic byproduct of ammonia, causes saponification of the lipid layer of cell membranes with consequent liquefactive necrosis and release of various proteins normally sequestered in subcellular compartments. Ammonium hydroxide also produces alkaline proteinases, which permanently alter the structure of proteins. Both released and altered proteins may function as autoantigens. Professional (polymorphonuclear leucocytes, monocytes) and nonprofessional (keratocytes expressing class II major histocompatibility complex) antigen-presenting cells (APCs) have both been found in alkali-burned corneas very shortly after the injury. Also, formation in the cornea of new blood and lymphatic vessels has been shown following alkali injuries. Interestingly, various circulating autoantibodies toward denatured corneal proteins have been reported in rabbits 5 weeks after alkali burns. The influx of immune cells combined with the presentation by APCs of corneal autoantigens could be the underlying process leading to development of PUK in these patients. Detection of circulating autoantibodies against corneal antigen was not performed in our patients.
In all our patients, there was a disease-free interval of several years between the alkali injury and PUK. The corneal autoimmune response may take a long period to develop and/or manifest clinically. A similarly protracted time lag occurs in the delayed-onset keratitis secondary to sulfur mustard gas exposure. Sulfur mustard is a vesicant agent consisting of an aerosol of oily droplets, which are rapidly absorbed by the ocular surface as well as other visible tissues. It causes inhibition of DNA replication and respiratory enzymes, ultimately leading to cell death. The severity of the acute ocular injury depends on the dose of exposure with keratitis, transient intraocular hypertension, anterior uveitis, and limbal ischemia characterizing the most severe cases. The acute lesions usually resolve within a few weeks. Residual corneal opacities and limbal stem cell deficiency may result from the acute exposure and cause chronic/recurrent sequelae.
In 0.5%–1% of all patients exposed to mustard gas, a delayed form of keratitis has been reported to develop 8–40 years after the initial exposures. It is characterized by recurrent episodes of limbal inflammation with associated peripheral keratitis with corneal neovascularization, lipid deposition, corneal stromal destruction, thinning, and, in some cases, perforation. Histologically, the disease closely resembles PUK. It is currently not known why only some of the eyes exposed to mustard gas develop the delayed-onset keratitis. The dose and modality of exposure as well as personal susceptibility may come into account. A local mustard gas–induced vasculitis or autoimmune process has been advocated in the pathogenesis. The clinical appearance, histology, and course of this disease are very similar to the cases hereby reported of delayed-onset alkali-induced peripheral keratitis. In both diseases, a chemical injury that occurred in the past might have triggered an autoimmune mechanism ultimately causing a late-onset PUK.
In conclusion, we report 5 cases of delayed PUK following alkali injury. To the best of our knowledge, this condition has not been described before. A putative autoimmune mechanism underlying the disease is proposed. Further studies are needed to clarify the pathogenesis and better define the therapeutic approach in this rare but sight-threatening condition.
All authors have completed and submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest and none were reported. Part of J.K. Dart’s salary was supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology. The views expressed are those of the author and not necessarily those of the NHS, the NIHR, or the Department of Health. Contributions of authors: design (A.I., J.K.D.); collection, management, analysis, and interpretation of the data (A.I., S.A., J.K.D.); preparation, review, and approval of the manuscript (A.I., S.A., J.K.D.).
Appendix: Case Reports of Patients With Peripheral Ulcerative Sclerokeratitis Associated With Alkali Chemical Burn
Case 1
A 22-year-old subject suffered a severe alkali injury with ammonia (hair dye) to his left eye in December 2007. There was complete loss of corneal epithelium with almost 360 degrees limbal ischemia. Complete re-epithelialization was achieved 3 months after the injury with 20/20 unaided visual acuity.
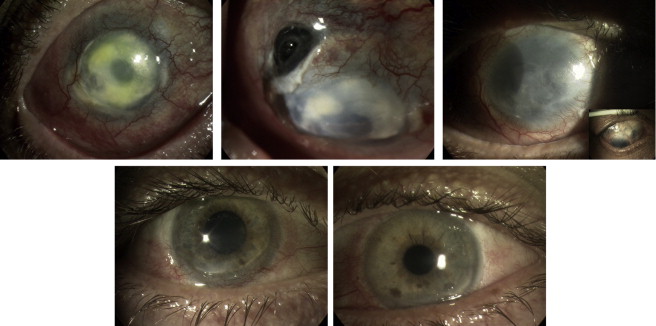
He presented to casualty 4 years later (October 2011) with a severe left peripheral ulcerative keratitis (PUK) involving 4 clock hours of the nasal cornea with severe limbal inflammation and peripheral corneal ulceration with 80% stromal thinning ( Supplemental Figure , Top left). The vision was 20/400 improving to 20/40 with pinhole. The patient was treated with pulsed intravenous methylprednisolone (1 g daily over 3 days) followed by 60 mg of oral prednisolone daily. Autoimmune disease screening was negative and no infection was detected on corneal scraping.
The peripheral corneal thinning progressed in spite of medical treatment, leading to descemetocele formation. A multi-layered amniotic membrane transplant (inlay and overlay) was carried out. Following this, the eye stabilized except for a small area of persistent epithelial defect at the edge of the amniotic membrane, which healed on autologous serum drops. Oral prednisolone was tapered.
In January 2012, there was recurrence of sclerokeratitis while the patient was on 10 mg of oral prednisolone, with a new broad area of peripheral ulcerative keratitis adjacent to the previously healed area and severe conjunctival inflammation ( Supplemental Figure , Top center). The vision was 20/200 improving to 20/60 with pinhole. Oral prednisolone was increased to 75 mg/daily and the patient was commenced on oral mycophenolate 1 g twice daily. Autoimmune screening with a rheumatology consultation was repeated at this point and no vasculitis or other autoimmune condition predisposing to PUK was found.
The patient stabilized on a high dose of oral steroids but the condition flared up again once the prednisolone was reduced to 15 mg/daily. Mycophenolate was increased to 1.25 g twice daily and oral cyclosporin A (75 mg twice daily) was added, along with an increase in oral prednisolone to 75 mg per day with gradual taper. However, the eye continued to be inflamed with circumferential progression of peripheral ulcerative keratitis and increasing central corneal haze.
At this stage the patient was referred to Moorfields Eye Hospital. The vision in the left eye was 20/200 improving to 20/60 with pinhole. Severe inflammation with superior and inferior nasal pseudopterygia was present. There was corneal stromal thinning (90%) nasally with epithelial defects ( Supplemental Figure , Top right). Neovascularization was present inferotemporally with lipid keratopathy. The central cornea had mid-stromal gray opacities that resembled an infectious crystalline keratopathy. Repeat corneal scrapings, corneal biopsy, and confocal microscopy did not show any sign of infection.
The patient was treated with intravenous infliximab in addition to the oral immunosuppression. On this regime the eye stabilized. By this time, there was almost 360 degrees of peripheral corneal thinning, scarring, and vascularization with a small central island of hazy but full-thickness cornea. The patient decided to discontinue immunosuppression after 6 months owing to repeated infectious episodes (perianal abscesses) leading to loss of earnings. At last follow-up, he had been off all treatment for over a year with no recurrence of sclerokeratitis. His cornea had stabilized with a visual acuity of 20/200 improving to 20/80 with pinhole, though there was diffuse scarring with superficial and deep vascularization and significant residual thinning.
Case 3
This 38-year-old patient suffered an ammonia chemical burn in both eyes (left more than right) in 1996. His past medical history was of left amblyopia and Crohn disease treated with oral mesalazine. The patient was referred to us for management of recurrent left keratitis starting in 2004, about 8 years after the initial injury. He had superonasal corneal pannus and lipid keratopathy associated with a central area of ulceration with mild stromal thinning ( Supplemental Figure , Middle left). The corneal ulcer was culture negative and thought to be consequent to stem cell deficiency secondary to the chemical burn, and it eventually healed with the aid of topical steroids and antibiotics. In March 2005 the patient was clinically stable with 20/30 vision and was discharged to follow-up with the referring ophthalmologist.
In late 2006 he again experienced several episodes of recurrent sclerokeratitis, resulting in a corneal perforation in March 2007, treated with application of Histoacryl glue (TissueSeal, Ann Arbor, Michigan, USA) and a bandage contact lens. The patient was reglued 2 more times until a large-diameter tectonic penetrating keratoplasty incorporating limbus with an amniotic membrane onlay was performed in April 2007 ( Supplemental Figure , Middle center). The patient was started on oral steroids and cyclosporin A to control the postkeratoplasty scleritis, which responded in about 6 weeks. The oral prednisolone and cyclosporin A were tapered and then suspended over a few months and the patient achieved a pinhole vision of 20/60. Cataract extraction was performed in his left eye in early 2008 with a postoperative vision of 20/40.
Recurrent epithelial breakdown with stromal loss ( Supplemental Figure , Middle right) reoccurred in 2009–2010 and was thought to be consequent to a combination of limbal stem cell failure and lagophthalmos, for which he underwent an upper lid levator botulinum toxin injection first and then an upper lid skin graft in April 2011. At the last assessment the vision was 20/120 with the aid of a diagnostic rigid contact lens and improving to 20/40 with pinhole.
Case 4
This 23-year-old subject was first seen in January 2011 at the Accident and Emergency Department at Moorfields Eye Hospital complaining of a 4-week history of red, painful, watery right eye with yellow discharge. The subject had no fixed address and was treated for depression with citalopram. He had suffered an alkali chemical trauma (ammonia) to the right eye 5 years prior. The right visual acuity was 20/200, not improving with pinhole. The right eye was inflamed with conjunctival hyperemia and chemosis. Corneal neovascularization was present with conjunctivalization superiorly and an inferior 1 mm peripheral keratitis extending from 3 to 9 o’clock with 70%–80% thinning ( Supplemental Figure , Bottom). Corneal scrapings were taken for cultures and the patient was started on topical dexamethasone and levofloxacin. A week later, the patient was reviewed in the clinic and did not show signs of clinical improvement; he was therefore started on a tapering course of oral steroids (prednisolone 80 mg daily). Corneal cultures as well as autoimmune screening were negative.
The patient missed several clinic appointments and was next seen 4 months later. At that time he appeared confused and repeatedly asked for psychiatric support. Compliance to topical or oral treatment could not be determined. On examination, the peripheral corneal melting had progressed circumferentially but the conjunctival and ciliary injection had improved. The patient refused hospitalization.
One month later, the patient came again to Accident and Emergency complaining of a recrudescence of red painful right eye. The patient reported that a botulinum toxin–induced ptosis had been performed on his right upper eyelid in another hospital, supposedly for an indolent corneal ulcer. He was not on any oral medications. His right visual acuity was hand motion. Circumferential PUK with moderate limbal inflammation was present; the central stromal cornea was diffusely hazy with deep stromal neovascularization temporally and nasally.
Owing to the patient’s lack of compliance, and for safety reasons, it was decided to use only topical treatment (dexamethasone and chloramphenicol).
Six months later the patient was reviewed in the clinic after missing repeated follow-up appointments. The right visual acuity was still hand motion. The patient was not on any topical or oral treatment. The right eye was now only mildly inflamed with 360 degrees of peripherally thinned cornea and a 4 × 4 mm central area of ulceration with stromal necrosis. The patient was started on topical dexamethasone and chloramphenicol. The option of a tectonic large-diameter lamellar keratoplasty combined with autologous limbal stem cell transplant and oral immunosuppressive treatment was discussed with the patient, but ultimately was not carried out owing to the patient’s loss to follow-up.