Histiocytic Disorders
This chapter includes non-neoplastic disorders featuring proliferations of histiocytes that may occur in any soft tissue or bony component of the body. Clinical entities of this group are Langerhans cell histiocytosis (LCH), sinus histiocytosis with massive lymphadenopathy (SHML), juvenile xanthogranuloma (JX), Erdheim-Chester (EC) disease, and sarcoidosis.
Langerhans Cell Histiocytosis
This entity includes three subtypes, the usually solitary eosinophilic granuloma of bone, the disseminated Schüller-Christian syndrome, and the disseminated fatal variety Letterer-Siwe disease, Unni (1996) seems reasonably sure that eosinophilic granuloma and Schüller-Christian syndrome represent stages of the same disease process, but it is possible that Letterer-Siwe disease is caused by a variety of conditions.
Incidence
In early literature, LCH was usually considered a disorder chiefly confined to children. However, Howarth et al. (1999) analyzed 314 Mayo Clinic patients with histologically proved LCH over a 50-year period. Forty-two percent (n= 131) were <20 years of age. Five patients (1.6%) had orbital proptosis. The period of this study was between 1946 and 1996, an interval almost the same as our 1948 to 1997 survey of orbital tumors. The most common sites of involvement were the calvarium, spine, and proximal femur.
Our 50-year survey of orbital LCH included 12 patients, 0.6% of the total 1,795 orbital tumors. In the orbital group, there were two unifocal, six multifocal, and four secondary tumors. The male to female ratio was 9:3. Seven patients on this list were 3 years or less of age. The two oldest patients were 17 years old. The Schüller-Christian syndrome and the unifocal eosinophilic granuloma most often affect children and young adults. The disseminated Letterer-Siwe usually affects very young children but can also occur in adults.
Clinical Features
Of most interest to the reader are the cases in which orbital symptoms alone, or associated with multifocal symptomatology, are the presenting features of the disorder. Some combination of unilateral proptosis and swelling of the lateral portion of the upper or lower eyelid is the most frequent initial symptom in these cases (see Fig. 14.1). These symptoms usually evolve over a period of 1 to 6 months. In general, the shorter the interval, the younger the child. In tiny tots, an accompanying redness and tenderness of the adnexal soft tissues suggests subacute cellulitis. Such cases are usually first treated with an antibiotic. In adolescents, tenderness is often absent. In such cases, onset is more protracted and the patient may not be brought for consultation until a reddish-brown or reddish-blue pustule-like lesion appears in the upper lid fold or the crease of the lower eyelid corresponding to the inferior orbital rim (see Fig. 14.2). Finally, when the child or adolescent does not respond to antibiotic therapy or conservative observation, computed tomography (CT) scan of the orbit reveals the defect in orbital bone. In our small series, the frequency of involvement of the zygomatic bone was nearly as great as that of the frontal bone.
In patients with multifocal disease, the presenting pattern is less specific. Here, we are often dealing with patients showing lytic defects of the sella turcica and sphenoid bone secondary to retro-orbital or orbital apex involvement. Such patients may show some measure of visual loss, afferent pupillary defects, painful ophthalmoplegia, sensory loss secondary to trigeminal nerve disease, otitis media, and stomatitis. Two of our patients developed the infrequently seen Schüller-Christian syndrome consisting of osteolytic lesions, diabetes insipidus, and exophthalmos.
In other interesting cases of LCH in recent literature, Kramer et al. (1997) reported three female children, aged 24, 24, and 21 months, respectively, in Nogales, Arizona, with involvement of the sphenoid bone and lateral orbit by tumor. The authors calculated an incidence rate of 40 per million, which is approximately 26 times the expected rate (P = 0.0001). This cluster of cases may imply that LCH may be a sentinel disease for unusual environmental exposure.
MacCumber et al. (1990) observed an adult patient who developed the chronic, diffuse type of tumor without osseous lesions over a 7-year period. An autopsy showed tumor spread to the dura, sagittal sinus, pericardium, both orbits, sella turcica, sclera, optic nerve, and choroid. Stromberg et al. (1995) noted a 16-year-old boy with a biopsy-proved mass in the sphenoid sinus that secondarily produced destruction of the lateral wall of the sinus and orbital invasion.
MacCumber et al. (1990) observed an adult patient who developed the chronic, diffuse type of tumor without osseous lesions over a 7-year period. An autopsy showed tumor spread to the dura, sagittal sinus, pericardium, both orbits, sella turcica, sclera, optic nerve, and choroid. Stromberg et al. (1995) noted a 16-year-old boy with a biopsy-proved mass in the sphenoid sinus that secondarily produced destruction of the lateral wall of the sinus and orbital invasion.
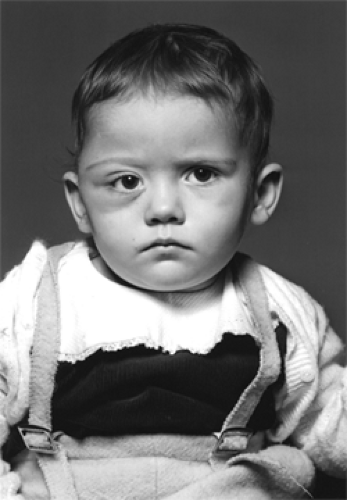 Figure 14.1 Langerhans cell histiocytosis: Swelling of the right upper face with downward and inward displacement of the eye secondary to lytic defects in the zygoma and lateral wall of orbit with extension of soft tissue component into the orbital cavity. This 2-year-old child eventually died from disseminated disease. |
Imaging Aspects
CT scan appearance is quite distinct. In the early stages, the lesion appears as a sharply delineated, radiolucent area of expanding bone. When the soft tissue component of the lesion breaks through the thinned-out cortex, the osseous defect has a dished-out, somewhat moth-eaten appearance (see Fig. 14.3). The bony lesion may be solitary in the orbit, or the calvarium will show other lytic defects (geographic skull). The soft tissue component usually shows some contrast enhancement. These bony defects heal if the child does not succumb to the systemic and visceral manifestations of the disease. In the healing process, the affected bone may undergo sclerosis with ultimate reduction in the overall size of the orbit. This accounts for the residual prominence of the eyes of some children whose localized disease has been arrested. This sclerosis occurs in both treated and untreated patients.
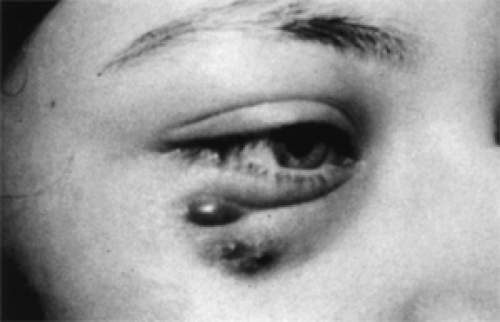 Figure 14.2 Nasal displacement of eye by Langerhans cell histiocytosis. The right lower eyelid had a reddish discoloration with fluctuant, purple, ecchymotic areas in the subcutaneous tissue that waxed and waned over a period of 1 month because of recurrent hemorrhage from underlying tumor. The tumor involved the inferolateral orbital wall with encroachment of a tender and indurated mass into orbital cavity. This 17-year-old girl was treated with irradiation. |
Pathology
Grossly, these tumors have been described as tan gray, tan yellow, yellowish red, and white in color. The hues of red probably reflect the vascularity of the lesion. The yellow
color probably indicates the amount of lipid accumulation in the lesion. The lesions are soft, friable, and often hemorrhagic.
color probably indicates the amount of lipid accumulation in the lesion. The lesions are soft, friable, and often hemorrhagic.
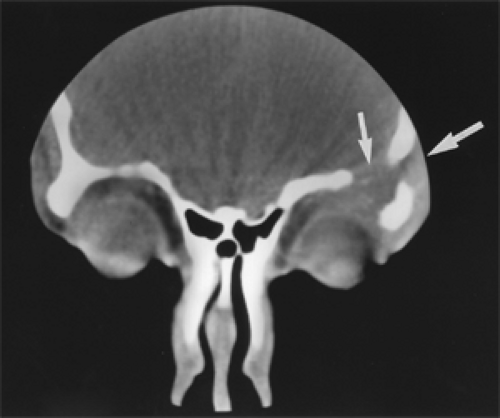 Figure 14.3 Coronal computed tomography scan shows lytic defects (arrows) in the left supraorbital ridge and orbital roof with slightly enhancing soft tissue extension into the orbit and adjacent anterior cranial fossa of a 16-year-old boy. |
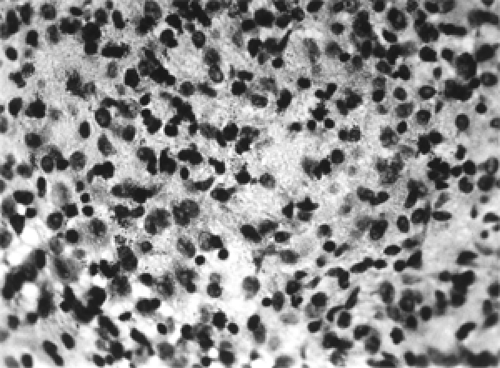 Figure 14.4 An admixture of histiocytes with ill-defined cell boundaries and finely vacuolated cytoplasm, and eosinophiles (× 400). |
The cellular component common to the localized, diffuse, and disseminated forms of LCH is the histiocyte. These histiocytes are usually arranged in loose sheets; have ill-defined cytoplasmic boundaries; have a pale, slightly foamy cytoplasm; and contain an oval or indented nucleus (see Fig. 14.4). Occasionally, the histiocytes have a focal arrangement and are completely surrounded by an abundance of eosinophiles. Inflammatory cells such as lymphocytes, plasma cells, and neutrophils are less in number. Multinucleated giant cells are frequent (see Fig. 14.5A and B). The histiocytes stain positive for S-100 protein and vimentin. The cell membrane receptors stain with T-6 monoclonal antibodies. Ultrastructurally, cytoplasmic inclusions, Birbeck granules, may be present in about 50% of cases.
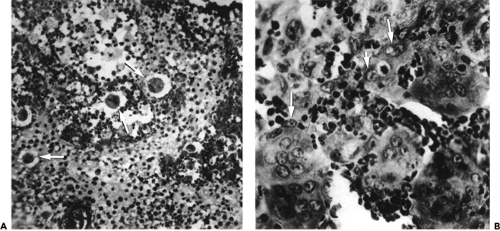 Figure 14.5 Histopathology A: Shows numerous histiocytes with pale cytoplasm (arrows) (× 250). B: An admixture of histiocytes with folded or indented nucleus membranes (arrows), multinucleated giant cells, eosinophiles, and lymphocytes (× 400). Both specimens from a 17-year-old girl with unifocal, orbital bone disease. |
Management and Course
Our small cohort of patients, mostly children (n= 12, age range 1 to 17 years) received three initial types of therapy over the 50-year period of study. Six patients, with focal lytic defects in the calvarium and skeletal bone, including the orbit, were treated only with irradiation. Three of these patients died 6 months to 3 years after therapy. The other three patients were alive 7, 14, and 29 years after initial treatment. Three of the patients had only lytic defects limited to the orbit. These patients underwent surgical removal of the soft tissue component of the tumor followed by irradiation. All are living 3, 6, and 28 years after treatment. One patient, a 17-year-old boy, received irradiation and andirine (surinamine) therapy and was living 15 years later. Two of our patients were lost to follow-up. There were no clues, initially, that we could use to predict which patients would or would not die except, possibly, the fewer the lytic spots treated, the longer the survival.
In the time interval of our tumor study, other treatments of LCH in the literature were surgery, irradiation,
and chemotherapy alone or in some combination, oral corticosteroids, intralesional injection of corticosteroids, low-dose radiotherapy, bone marrow transplantation, antibody therapy, intramuscular injections of an extract of thymus gland (a T-cell suppressor), and simple observation. We are not aware of any published protocol for the therapy of LCH, derived from a grouped medical center study. With the passage of time, we are more hesitant to recommend radiotherapy for young children. Instead, we favor a more conservative therapy, one of the above, as the initial remedy for LCH limited to the orbit.
and chemotherapy alone or in some combination, oral corticosteroids, intralesional injection of corticosteroids, low-dose radiotherapy, bone marrow transplantation, antibody therapy, intramuscular injections of an extract of thymus gland (a T-cell suppressor), and simple observation. We are not aware of any published protocol for the therapy of LCH, derived from a grouped medical center study. With the passage of time, we are more hesitant to recommend radiotherapy for young children. Instead, we favor a more conservative therapy, one of the above, as the initial remedy for LCH limited to the orbit.
Recently, a relatively new treatment looms on the horizon in treating disseminated LCH. This is a purine nucleoside analog, 2-chlorodeoxy-adenosine (2-CDA). Several reports of this drug have appeared in the literature since 1993. A more recent report is that of Pardanani et al. (2003). They treated five patients between December 1994 and January 2001. The age range of patients was 19 to 81 years. Median follow-up after initiation of treatment was 33 months. In one patient, the drug was used as frontline therapy and as salvage therapy for the other patients. Complete response was achieved in three patients and a partial response in the other two patients. The drug is a potent immunosuppressive and needs “to be studied prospectively.”
Eosinophilia is a major component in the orbital lesions of patients with LCH. When the eosinophil degranulates, it releases a major basic protein, indicating an effector role in the pathogenesis of this disease (Trocme and Aldave, 1994).
Sinus Histiocytosis
The disorder received its first name, SHML, and its stature as a clinicopathologic entity, chiefly through the publications of Rosai and Dorfman (1969, 1972). The disease consists of a proliferation of benign histiocytes with an admixture of lymphocytes chiefly in sinusoidal spaces of the cervical lymph nodes, resulting in their enlargement. Subsequently, it was noted that some histologically proved cases in extranodal sites had little or no cervical lymph node enlargement, which suggested that the original name was no longer appropriate. Consequently, the name was shortened to sinus histiocytosis (SH). The etiology of the disorder is not known.
Incidence
The primary locus of the disorder is massive and is a uni- or bilateral cervical lymphadenopathy. All ages may be affected, but there is a strong predilection for children. Extranodal sites are not uncommon and may be observed in about 43% of cases (Foucar et al., 1990). In the skull, the nasal fossa and paranasal sinuses are the most common extranodal sites. The orbit and central nervous system are both less common sites for manifestations of the tumor. Of the 423 patients in the SH registry, 36 (8%) had some evidence of orbital involvement (Resnick et al., 1996). Including all loci of the disorder, there is a mean age of presentation of approximately 20 years with a slight male predominance of 58%. We have not encountered a patient with SH in our 50-year file of orbital tumors.
Clinical Features
The cardinal feature of orbital presentation is a slow, painless, proptosis of one or both eyes over a period of several months. Associated signs and symptoms may include fever, leukocytosis, elevated sedimentation rate, hypergammaglobinemia, transient visual loss, and blindness. Blindness occurred in two 12-year-old patients of Remadi et al. (1996) because of massive tumors in the retrobulbar space that exerted such pressure and pain on the eye as to warrant enucleation. Other authors who have reported cases of orbital involvement in the literature in the past 15 years are Resnick et al. (a 38-year-old man), Burton et al. (a 2-year-old girl), Brau et al. (1995) (a 5-year-old boy), and Salmon and Duffield (1989) (a 12-year-old girl). Cases of bilateral orbital involvement are also reported. Optic nerves are rarely involved except by orbital compression from a massive tumor.
Imaging Aspects
The orbital image of this soft tissue mass with CT scan shows a homogeneously enhancing mass with a density and contour likened to lymphoma (see Fig. 14.6A). With magnetic resonance (Fig. 14.6B), the tumor is isointense to gray matter on all sequences (Burton et al., 1989). With increasing bulk, the orbital lesion may cause expansion of the foramina and fissures into adjacent paranasal sinuses or intracranial cavity, but bone destruction is seldom seen.
Pathology
Grossly, this tumor is whitish gray, firm, and usually adherent to surrounding orbital structures. However, the histology of the lesion is its most distinctive feature. Cytoplasmic vacuoles within the macrophage contain T-cell lymphocytes and other inflammatory cells—a phenomenon known as emperipolesis (see Fig. 14.7A). The outline of some cells is indistinct, but the nuclei are oval or round with regular outlines. The histiocytes stain positively with S-100 protein but do not contain the cytoplasmic organelle (Birbeck granules) peculiar to the Langerhans histiocyte. Fibrosis is a common component of most tissue specimens (Fig. 14.7B) and probably represents the natural course to involution.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


