Hematopoietic Tumors
In this chapter, we will discuss a select group from a wide array of hematopoietic neoplasms that, in their extranodal, extralymphatic, intravascular, and extramedullary forms, often affect the soft tissues of the orbit. This select group includes the non-Hodgkin lymphomas, the myelogenous tumors, and Hodgkin lymphomas. Also, we will add a brief discussion of the non-neoplastic lymphoid hyperplasia that, if the antigen stimulus responsible for the hyperplasia does not subside, may become malignant. Likewise, amyloidosis (AL), as it affects the orbit, is included because this protein dyscrasia is sometimes associated with a malignant clone of plasma cells.
Classification
Classification of these tumors, which is important for their diagnosis by the pathologist and for an estimate of their prognosis by the clinician, has been an integral component of the prior editions of this text. Starting with the decade of the 1940s and continuing through 1993, hematopoietic tumors were the subject of numerous classifications such as: Rappaport, Dorfman, Kiel, Lukes and Collins, World Health Organization (WHO) (1982), British National Lymphoma Investigation, Working Formulation, Revised European American Lymphoma (REAL), and non-Hodgkin Lymphoma Pathologic Project. However, there was considerable duplication inherent among the classification and the goal of a single worldwide classification encompassing the advances in diagnostic histopathology, genetic features, management, and survival of patients with hematopoietic tumors remained elusive. Accordingly, the WHO, in 1995, launched a project to provide “the first, true worldwide consensus classification of haematologic malignancies” that would include standardized nomenclature for the study and definition of the tumors’ clinical oncology, and multicenter therapy trials and comparative studies in different countries. They solicited proposals and advice from hematopathologists, geneticists, and clinicians. The classification project was completed in 2001 with their publication, Pathology and Genetics of Tumors of Haematopoietic and Lymphoid Tissues. This publication “stratifies neoplasms primarily according to lineage: Myeloid, lymphoid, histiocytic/dendritic cell and mast cell. Within each category, distinct diseases are defined according to a combination of morphology, immunophenotype, genetic features, and clinical syndromes.” In the latter category, a total of 75 distinct entities are listed. The group of mature B-cell neoplasms contains the largest (16) number of subtitles. The group of myelodysplastic/myeloproliferative disease and the group of immunodeficiency associated lymphoproliferative disorders each have the fewest (four) entities. The nomenclature for each entity reflects the editors’ best estimate of its lineage and stage of differentiation. Overall, this new classification seems very complex and may require several years before its practical value is proved. We are yet to convert the 175 hematopoietic tumors in our 50-year collection to the WHO classification.
Mature B-Cell Neoplasms
The title of this section was chosen by the WHO for the largest family of neoplasms within the wide array of hematopoietic tumors (see Table 13.1).
In the list (Table 13.1), we will first discuss the lymphomas that are known to involve the orbit, either as a localized tumor or part of the systemic disease. In prior editions of this text, these tumors were called non-Hodgkin lymphomas.
Non-Hodgkin Lymphoma
All of these tumors, despite the implication of benignity by the suffix “oma,” are malignant, with varying degrees of morbidity and mortality. These neoplasms do not, necessarily, have a cell of origin but are thought to be clonal proliferations of B cells at various stages of differentiation.
Incidence
These lymphomas are the most common malignant neoplasms associated with orbital involvement. In our 50-year collection, there are 175 lymphomas comprising 9.7% (175 out of 1,795) of the total tumors. Eighty-seven tumors,
4.8% of total tumors, were an extranodal manifestation of multifocal disease; 82 tumors, 4.5% of total tumors, were localized to the orbit; and six neoplasms (secondary type) invaded the orbit from a nasal, paranasal sinus, or conjunctival source—mucosa-associated lymphoid tumors (MALT). Almost all lymphomas with orbital involvement occur after the fourth decade. Only seven patients overall were <40 years of age. There was a slight preponderance of males (94) over females (81). Bilateral orbital involvement is relatively more frequent in the multifocal group. This incidence data does not apply to Burkitt lymphoma and large B-cell lymphoma because they occur more frequently in children.
4.8% of total tumors, were an extranodal manifestation of multifocal disease; 82 tumors, 4.5% of total tumors, were localized to the orbit; and six neoplasms (secondary type) invaded the orbit from a nasal, paranasal sinus, or conjunctival source—mucosa-associated lymphoid tumors (MALT). Almost all lymphomas with orbital involvement occur after the fourth decade. Only seven patients overall were <40 years of age. There was a slight preponderance of males (94) over females (81). Bilateral orbital involvement is relatively more frequent in the multifocal group. This incidence data does not apply to Burkitt lymphoma and large B-cell lymphoma because they occur more frequently in children.
Table 13.1 World Health Organization Classification | |
|---|---|
|
Clinical Features
In patients with unifocal orbital lymphomas, the single most common presenting sign is either eyelid-related or some protrusion or change in the position of the eye. Less common presenting signs in order of frequency are a visible mass, diplopia, epiphora, redness of the affected eye, and a foreign body sensation. The principal sign is a painless puffiness, swelling, or edema of an eyelid on the affected side. If an upper eyelid is affected, it may be “droopy” (see Fig. 13.1). Seldom is the affected eyelid red or congested in appearance. These eyelid manifestations seem related to the juxtaposition of a mass either in the anterior orbit just behind the orbital septum or in the conjunctival fornix. A mass in the conjunctival fornix will have a smooth surface and a distinctive red-salmon color (see Fig. 13.2).
Proptosis, if present, is seldom marked in degrees. Approximately 70% of patients will have 6 mm or less of proptosis on the affected side. Approximately 10% of patients may have no proptosis. Others may have only a slight displacement of the eye without forward protrusion, which reflects the presence of a mass in the far forward region of the orbital space.
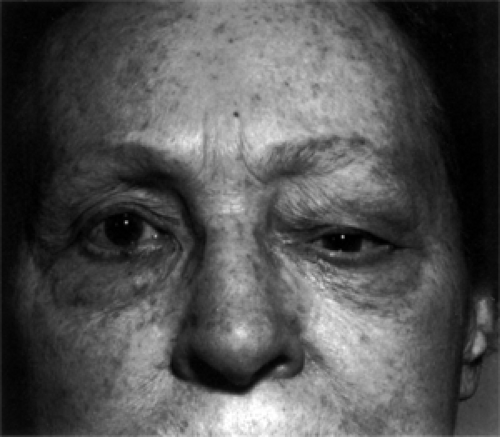 Figure 13.1 Lymphoma: A common presentation is a painless, unilateral swelling of an upper eyelid in an elderly individual. The droopy left upper eyelid of this patient conceals a slight downward displacement and minimal proptosis of the left eye. |
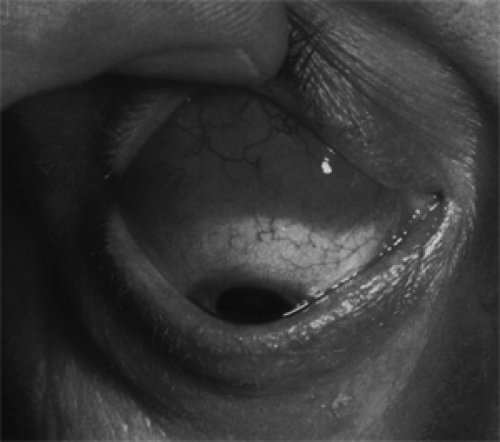 Figure 13.2 Lymphoma: A reddish salmon-colored mass was found on the surface of the right eye upon elevation of the upper eyelid of a 52-year-old man who had noted a slight prominence of the eye of 3 months’ duration. The mass was a forward extension of another palpable mass in the anterosuperior orbit. The pattern of blood vessels on the surface of the mass, its color, and tendency of tumor to adapt to the curvature of the eyeball are quite distinctive of lymphoma. (See Color image.) |
The onset of the disease is insidious, and the mean duration of the presenting complaint in our series was 10 months (range 1 to 48 months). Most patients will have two or more of the aforementioned signs and symptoms at presentation. Other than the color and contour of a mass in the conjunctival fornix, if present, the presenting features are not specifically relative to other orbital tumors in adults (see Fig. 13.3A).
None of the other major orbital tumors of adults are so often associated with a palpable mass as these lymphomatous tumors. This reflects the tendency of these soft tumors to extend along fascial planes of the anterior orbit, extraocular muscles, and surface of the eyeball, rather than forming an isolated circumscribed mass in the posterior orbit. In this sense, the tumors tend to conform to the space in which they grow. In the inferior orbit, where there is less fascial restraint to expansion, the tumors may feel “shoddy,” “corded,” “nodular,” or even “rubbery.” In the superior orbit, where they may lie just behind the orbital septum, the mass is firm and less easily defined. Approximately 83% of our patients had a palpable mass.
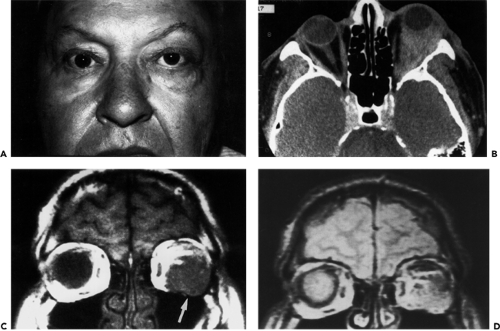 Figure 13.3 Lymphoma: A: A 61-year-old man with swelling of the temporal portion of the left upper and lower eyelids and slight upward displacement of the left eye. The edematous eyelids conceal a salmon-colored flat contoured mass overlying the left lateral rectus muscle and a purplish discoloration of the inferotemporal conjunctival fornix. B: Axial computed tomography scan showing the left orbit almost completely filled by a slightly enhancing homogeneous mass. The large size of the mass is disproportionate to the minimal objective findings in (A), a discrepancy not seen in most orbital tumors of adults. C: Magnetic resonance T1-weighted coronal scan of the same patient showing a homogeneous hypointense mass (arrow) relative to the signal intensity of orbital fat. D: The lesion as seen on the spin-echo sequence. |
The tumors may grow in any area of the orbit. In two of our patients, the tumors could be palpated in all four orbital quadrants. In general, these tumors localize in the superior orbit in a ratio of 2:1 over other combined orbital sites. The superior nasal and superior temporal quadrants are equally affected. In the superior nasal quadrant, the tumors are prone to cluster around the trochlea.
Other manifestations of these lesions such as significant visual loss, afferent pupillary defect, papilledema, and choroidal folds were seldom encountered. When present, these signs and symptoms usually indicate a lymphomatous tumor that has been unable to escape the more rigid fascial restraints at the orbital apex.
Patients with either known or unsuspected multifocal disease differ little from those with unifocal orbital lymphoma in their orbital presentation.
Imaging Aspects
With computed tomography (CT) scan, the tumors are relatively high-density masses that show definite but mild contrast enhancement and are not associated with bone erosion unless they are located in the peripheral orbital space, the lacrimal gland, or are of the secondary type. The lesions are usually homogeneous, and the imaging shadow has a soft putty-like appearance. However, these features of the scan are nonspecific. A clinical aspect is the tendency of the lesion to extend along fascial planes and contour around solid structures such as the eyeball and intraorbital optic nerve. In areas where the mass pushes against soft tissue structures of greater density, the border of the lesion appears circumscribed; where there is little resistance to expansion, the border of the mass looks fuzzy. If the lesion is limited to the peripheral orbital space, its border may appear angulated. Another interesting facet involves the patients with minimal proptosis who have a large bulky mass that fills the entire orbital space (Fig. 13.3B). This disproportion between orbital mass and displacement of the eye is due to the soft consistency of the tumor. Study of the lesion with magnetic resonance imaging (MRI) usually shows a hypointense mass relative to orbital fat and an isointense mass relative to brain (Fig. 13.3C and D) on T1-weighted sequence.
Iridium 111, a γ-ray emitter, can be used for radio-immunoscintigraphy as a surrogate of yttrium 90 monoclonal antibody, a β-ray emitter.
Pathology
The gross appearance of extranodal, unifocal orbital lymphomas gives no clue about their morphologic or immunotyping status. In color, the orbital tumors are some shade of gray, gray blue, tan, or reddish gray, which helps delineate the lesion from the surrounding yellow-hued orbital fat. Some observers have also described the tumors as purplish or maroon and likened this to the color of a cavernous hemangioma. In consistency, they are soft and friable but not necrotic. Although slightly lobulated, the lesions are reasonably compact when situated in the anterior orbit. Their surface is slightly circumscribed where the tumor abuts the orbital septum or is confined by a fascial partition. The posterior border of such a lesion, however, is less structured, infiltrative, and ill defined. They are reasonably avascular-appearing tumors until touched or manipulated. Then the surgeon is aware of the vascularity of their interlobular septae. When the tumors extend into the conjunctival fornices, their color changes to a reddish tan or salmon hue, and their smooth surface is covered by a visible vascular arcade. With low-magnification light microscopy, the tumors are hypercellular and stromal components are either absent or sparse. Two types of B-cell neoplasms comprised most of the unifocal, orbital lymphomas in our 50-year study. These were the small lymphocytic lymphoma and the follicular lymphoma. Occasionally, a third type was encountered, the mantle cell lymphoma. The first two are considered low-grade (indolent) neoplasms, whereas the mantle cell lymphoma is now known to be more aggressive (Looi et al., 2005). The small lymphocytic lymphomas are a diffuse proliferation of lymphocytes of nearly normal size with a scanty cytoplasm, round or dark nuclei, coarsely clumped nuclear chromatic, and inconspicuous nucleoli interspersed in a sparse stroma (see Figs. 13.4 and 13.5).
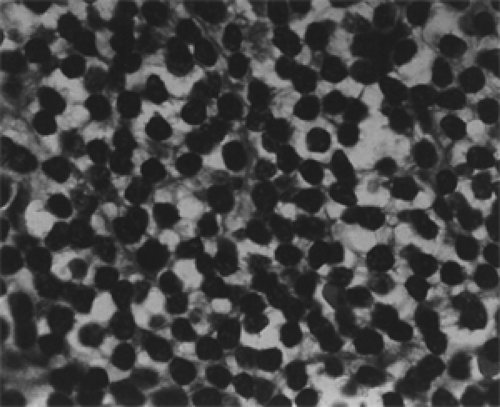 Figure 13.4 Lymphoma: Small lymphocytic diffuse type showing a uniform population of round and oval cells with regular nuclear outlines and scanty cytoplasm within a delicate stroma (× 800). |
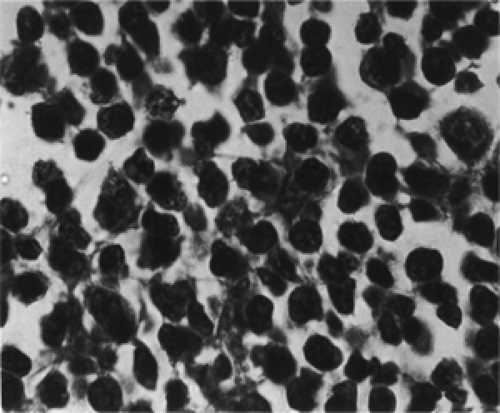 Figure 13.5 Lymphoma: Mixed type showing a population of small and large cells with differing amounts of nuclear chromatin and cytoplasm (× 800). |
 Figure 13.6 Follicular lymphoma: Ill-defined follicles without a clear distinction between mantle zones and follicular center zones. In both zones, the same monoclonal, cleaved cells are present (× 100). (From Spencer WH. Follicular lymphoma. Ophthalmic Pathology. 1996;4:2710. ) |
In the follicular lymphoma, the germinal center with its surrounding zone of small lymphocytes (normal follicle) is replaced by a collection of small monoclonal lymphocytes with cleaved (infolded) nuclei. In the mantle cell lymphoma, the architecture of the follicle is preserved, but the periphery consists of monoclonal, small lymphocytes (see Figs. 13.6, 13.7, and 13.8).
Immunophenotypes
This staining modality is a useful diagnostic adjunct to differentiate benign lymphoid hyperplasias from lymphomas and, in addition, to separate some lymphoma types from one another. In general, the B-cell lymphomas of this section are positive for surface immunoglobin M (IgM), IgM and IgD, CD5, CD19, CD20, CD22, CD79a, CD23, CD43, CD11c, and as a rule, are negative for CD10, cyclin D1, and CD76b. CD23 and cyclin D1 are useful in distinguishing the leukemic and soft tissue phases of small cell lymphoma from mantle cell lymphoma. Rare cases of mantle cell lymphoma may partially express CD23; therefore, cyclin D1 should be assessed in lymphomas that are CD+ and CD– (WHO classification).
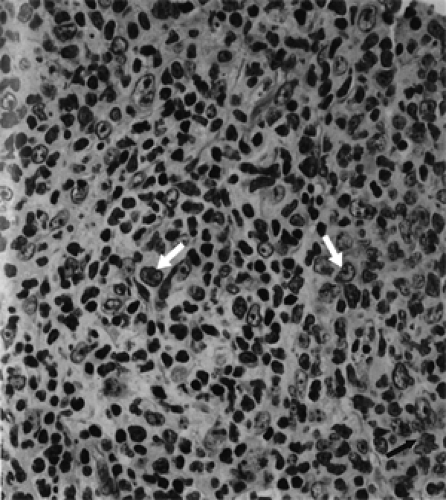 Figure 13.7 Lymphoma: A diffuse proliferation of predominantly small lymphoid cells which show (arrows) marked nuclear angulation and indentation (cleaved cells) (× 640). |
 Figure 13.8 Mantle cell lymphoma: Small neoplastic cells proliferate as large, expanded mantles around residual benign germinal centers (× 80). |
Management and Course
Esik et al. (1996) published a retrospective review of experience at a single institution with 37 patients having “primary,” orbital lymphoma treated with three modalities: Radiotherapy (17 cases), surgery alone (13 cases), and chemotherapy (7 cases). Patients were followed up with a mean and median of 7.6 and 6.2 years. There were 34 low-grade tumors; small lymphatic lymphoma, follicular lymphoma, and mantle cell lymphoma. The remaining three were higher grade lymphomas. Only patients with low-grade tumors were analyzed for effectiveness of treatment, recurrence, and survival.
Briefly, a summary of their conclusions were:
Radiotherapy in a relatively low dose (30 Gy) has the most decisive impact on the tumor when administered as the initial treatment. There was complete remission of the lymphoma over a 10-year period.
There was a high relapse rate in patients after surgery alone. Radiotherapy, at the time of relapse, was given to bring about remission.
Initial treatment with multiagent chemotherapy showed only a very slow, and sometimes no effect. It was difficult to achieve complete remission, even when radiotherapy was also given. Survival may be worse after initial chemotherapy (even with salvage radiotherapy) than with initial radiotherapy.
For intermediate and high-grade lymphomas, chemotherapy in combination with radiation therapy was shown to be more effective than chemotherapy alone (Miller et al., 1998). Subsequently, advances in biotechnology have led to the creation of a biologically active, unlabeled anti-CD20 chimeric (murine-human) antibody called rituximab. This is the first monoclonal antibody licensed (November 1997) by the Food and Drug Administration after review of multi-institutional, pivotal study of 166 patients confirmed its antitumor activity in the treatment of relapsed, low-grade lymphomas, particularly follicular lymphomas. The target for this antibody is the CD antigen. This antigen, on the surface of the cell, is present in >90% (Leget and Czuczman, 1998) of the family of B-cell lymphomas of our present regard.
We found only one report of the use of rituximab for low-grade lymphoma of the orbit (Esmaeli et al., 2002). Three patients received intravenous rituximab 375 mg per m2 weekly for 4 weeks. The patients had nearly complete resolution of the orbital lymphoma in a follow-up time from 6 to 22 months (mean 14.5 months). The most common adverse effect is B-cell depletion. The authors believe targeted immunotherapy may offer several advantages, in the future, over conventional chemotherapy or external beam radiotherapy in the treatment of orbital lymphomas.
Most hematopoietic malignant neoplasms are radiosensitive, particularly the lymphomas. Iodine 131-radiolabeled antibody has been used in the past for relapses of low-grade lymphoma and initial therapy of high-grade malignant tumors. More recently, the β-emitting isotope yttrium 90 has received favorable reviews for its effectiveness, as an alternative to iodine 131, in the management of lymphomas. Yttrium 90 emits β rays that are five times as energetic as those from iodine 131 (Press et al., 1999). Also yttrium 90 emits very few γ particles and has a favorable half-life of 2.5 days. It is used on an outpatient basis. Anti-CD20 antibodies radiolabeled with iodine 131 and yttrium 90 administered at nonablative doses yield remission in 75% to 80% of cases, including 35% to 40% complete remissions (Kaminski et al., 1993). “Higher-dose radio-immunotherapy combined with stem cell transplantation also induces responses in >85% of patients, most of whom achieve complete remission. Encouraging trials have also been conducted with antibodies targeting other B-cell antigens, including immunoglobulin isotopes CD22” (Press, 1999).
Summary
Immunotherapy is still so new, the agents recommended for therapy so diverse, a sizable collection of cases of a particular type of lymphoma and its response to therapy so small, follow-up periods so short, that neither a standardized protocol for therapy nor an estimate of survival has evolved. Reports of immunotherapy for localized orbital lymphoma are very few.
In our long association with a collection of consecutive, pathologically-proved cases of orbital tumors, radiation has been the mainstay of initial therapy for lymphomas localized to the orbit. Stafford et al. (2001) attempted an update of patients treated from 1971 to 1987, inclusive, as well as a longer follow-up on Mayo patients described by Minehan et al. (1991) Their analysis was limited to 46 patients, 45 with low-grade and 1 patient with intermediate-grade histologic findings based on the working formulation and, later, the REAL classification. This cohort, if converted to the present WHO classification, would consist of 29 extranodal marginal zone B-cell lymphomas of MALT, 11 small lymphatic lymphoma, and 6 follicular lymphomas. About half of this total was localized to the orbit; the remainder involved conjunctiva and lacrimal gland.
Radiation doses ranged between 15 and 53.8 Gy (median 27.5 Gy). Acute complications occurred in half of the patients but resolved by the first follow-up visit. Conjunctivitis was the principal acute complication. Severe complications occurred in four patients, that is, cataract, sicca syndrome, keratitis, and mild radiation retinopathy. No late complications occurred with doses <35 Gy. Lens-blocking techniques did not seem to have any significant effect on the reduction of cataract in patients aged 68 years or older.
The 5- and 10-year rates of survival were 69% and 40%, respectively, for the entire cohort of patients. The authors believe control of local orbital lymphoma is possible with moderate-dose radiation therapy in tumors of low histologic grade, including MALT lymphomas. As of 2004, radiotherapy is still the preferred modality for the initial management of orbital lymphomas. The radiation dosage has been reduced to 25 Gy at the Mayo Clinic in an effort to further minimize post-radiation complications to the eye. An occasion appropriate for the use of rituximab has not occurred in these patients, although rituximab is used for the treatment of some types of systemic lymphoma.
Burkitt Lymphoma
This is a high-grade B-cell neoplasm. It is usually found in the pediatric population. Many pediatricians consider the tumor as the most aggressive malignancy in this age-group. It exists in endemic, sporadic, and human immunodeficiency subtypes. The endemic type is better known as the African subtype. Names for the sporadic form are: North American type, nonendemic Burkitt lymphoma, and non-African Burkitt lymphoma. It usually presents as a multifocal neoplasm; although rarely, it may apparently
be localized to one anatomic site such as the orbit. The histopathology of the tumor is the same regardless of its geographical location.
be localized to one anatomic site such as the orbit. The histopathology of the tumor is the same regardless of its geographical location.
Clinical Features
The African neoplasm is virtually always associated with the (herpes-like) Epstein-Barr virus (EBV). Positive titers are found in over 90% of cases (Davi et al., 1998). It is diagnosed at an average age of 9 years (Banthia et al., 2003). These children present with a rapidly growing mass (see Figs. 13.9 and 13.10) in the maxilla or mandible. The maxilla is affected more often than the mandible in a ratio of approximately 2:1. As the tumor expands, the teeth on the affected side become loose; there is odontalgia, paresthesias of the cheek, sore throat, enlarged cervical nodes, fever, and secondary orbital encroachment. In the meantime, an enlarging abdominal mass may go unnoticed. If the disease goes untreated, soon there is invasion of bone marrow, dissemination to the lungs and the central nervous system, and extension of tumor into the posterior orbit. Ocular motility disturbances and palsies of cranial nerves III and VI are late manifestations of either orbital or brain involvement.
The sporadic type is more heterogeneous, and an association with EBV is less often detected. Also, there is more frequent initial presentation of abdominal and mediastinal masses and lymph node and bone marrow involvement. The disease tends to occur in an older age-group than in the endemic African type, and jaw involvement is seen in only a minority of cases (12%) according to the American Burkitt Lymphoma Registry (Yih et al., 1990).
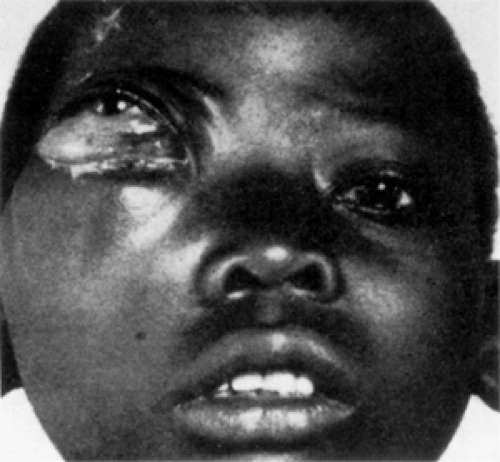 Figure 13.9 Burkitt lymphoma: One of the 38 African children described by Dr. Denis Burkitt with an evolving sarcoma of the jaw. The tumor originated in the right maxilla of a 10-year-old boy and extended into the orbit. The tumor did not arise in relation to the teeth. (From Burkitt D. A sarcoma involving the jaws in African children. Br J Surg. 1958;46(197):218–223 . By permission of the publisher Butterworth-Heinemann, Ltd.) |
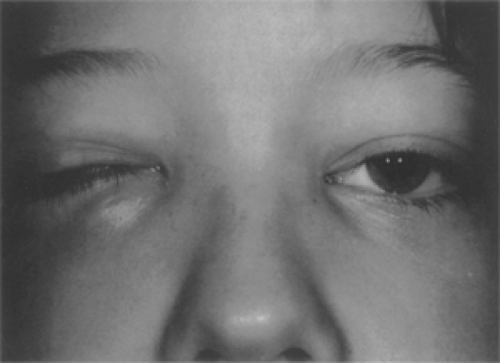 Figure 13.10 A 9-year-old boy whose assumed orbital “cellulitis” was unresponsive to a month’s course of antibiotics. A computed tomography scan then revealed a mass in the inferonasal orbit that, on biopsy through the medial portion of the right lower lid, proved to be a Burkitt lymphoma. The patient died 2 months later. (Courtesy JD Bullock, Dayton, Ohio.) |
Weisenthal et al. (1995) reported a 16-year-old girl with a rapid development of a conjunctival mass 6 weeks after an illness associated with a positive Monospot test. CT scan showed extension into the anterior orbit. Evidence of EBV infection was not found in the tumor cells but the EBV-encoded nuclear RNA was present in the surrounding adenoid tissue. The patient was treated with combined chemotherapy and was disease free 4½ years after diagnosis.
Edelstein et al. (1997) describe a 26-month-old boy who developed oral thrush associated with painless swelling of the right eyelid, 10-mm proptosis, a diffuse orbital mass, and both maxillary sinuses. Abdominal CT scan showed involvement of the kidney, pancreas, and liver. Bone marrow biopsy disclosed a neoplastic clone of cells bearing the t(8;14) translocation. This patient received six cycles of combined chemotherapy with complete regression of both orbital and systemic manifestations in 8 months.
Another sporadic case is the 6-year-old girl with progressive right facial and orbital swelling with loss of vision in the right eye (Banthia et al., 2003). MRI revealed an orbital mass, maxillary tumor, and hard palate involvement. An extensive physical examination did not reveal systemic disease. Immunogenetic analysis revealed cells bearing the t(8;14) translocation. The patient was still undergoing chemotherapy at the time of their publication.
In our third edition (1994), we described five cases of sporadic Burkitt lymphoma. We will not redescribe these cases but will list the authors of these cases in our references (1978 to 1990) at the end of this chapter (Blakemore et al., 1983; Kielar, 1978; Trese et al., 1980; Zak et al., 1982).
Burkitt lymphoma also occurs in patients with acquired immunodeficiency syndrome (AIDS). This form of the tumor is similar to the preceding clinical type except for a higher initial presentation with central nervous system disease. A case of this type with unilateral orbital involvement was the 22-year-old man with a positive Epstein-Barr antigen titer, an elevated cytomegalovirus immunoglobulin titer, and inversion of the T-cell helper-to-suppressor ratio. On CT scan, the right globe was displaced by a large enhancing orbital mass (Brooks et al., 1984).
The imaging characteristics of these neoplasms by CT scan and MRI are essentially the same as described for the preceding non-Hodgkin lymphoma.
Histopathology and Immunocytology
The histopathologic pattern is a proliferation of small, closely packed lymphocytes interspersed with phagocytic histiocytes, which has been likened to a “starry sky” (see Fig. 13.11). The small lymphocytes have uniform oval or round nuclei with distinct nuclear membranes and contain one to several distinct basophilic nucleoli. Each cell has a rim of amphophilic cytoplasm. Mitotic figures are frequent. In contrast, the macrophages are very large with an abundant clear cytoplasm containing speckles of cellular debris. The intercellular reticulin network is delicate and sparse.
Immunocytochemical marker studies are positive for antibodies to leukocyte common antigen, B-cell lineage markers including C19, CD20, CD22, CD74, CD79a, and CD10. Also expressed are cell surface immunoglobulin heavy chains—most commonly IgM and either κ or λ light chains (Freedman and Nadler, 1991). Histiocyte markers KD-1 (CD68) stain the macrophages.
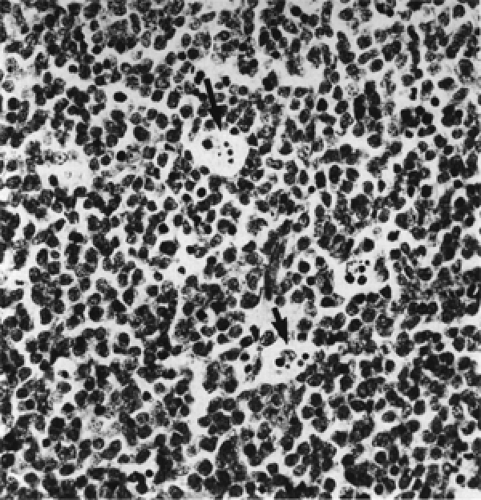 Figure 13.11 Burkitt lymphoma: Starry-sky appearance is due to large, pale histiocytic cells scattered among closely packed, small lymphocytes (× 400). Some histiocytes contain phagocytosed nuclear material in their cytoplasm (arrows). |
Treatment
The primary therapeutic modality for Burkitt lymphoma is chemotherapy. The neoplastic cells are highly sensitive to cytotoxic agents (Banthia et al., 2003). Most chemotherapy regimens are cyclophosphamide based. A combination of multiple agents with different mechanisms of action maximizes dose intensity and decreases the chance of drug resistance. Patients with Burkitt lymphoma limited to the head and neck have a 90% long-term survival when treated with multiple-agent chemotherapy regimens. Patients with involvement of bone marrow, brain, and HIV fare less well (Link et al., 1990). The central nervous system is a common site of relapse of disease.
Extranodal Marginal Zone B-Cell Lymphoma (WHO Classification)
This neoplasm is a relatively new member of the lymphoma family, considering the known, multidecade association of other small cell lymphomas with orbital involvement. Isaacson and Wright (1983) thought these neoplasms should be a separate entity from other small cell lymphomas because their origin was not in a lymph node but in the follicular lymphatic aggregates (Peyer patches) of the gastrointestinal tract—therefore, the name, extranodal. The authors proposed the term MALT.
Within a year, the concept of MALT lymphomas extended to extranodal sites other than gut (Banks and Isaacson, 1999). Soon after, the neoplastic element was identified as the B cell (memory cell) in the marginal zone surrounding the reactive (benign), antigen-processing center of the lymphoid follicle. Antigen dependence is still considered to be an important factor in the pathogenesis of the neoplasm. In brief, these neoplasms differ in morphology, immunophenotype, and clinical course from other small cell lymphomas.
Our interest is chiefly the MALT lymphoma of the extranodal lymphoid tissue of the conjunctival submucosa or the nonlymphatic reticulum in the posterior orbit. The incidence of the tumor in these two sites is not known because they are either lumped together with the incidence of head and neck lymphomas, or combined in a discussion of extraocular MALT lymphomas. Most MALT lymphomas occur in adults with a median age of 61 and a slight female preponderance (male:female ratio 1:1.2) (Jaffe et al., 2001). The conjunctival neoplasms are, by far, more common than orbital lymphoma.
Because the consistency of the MALT lymphoma is so soft and its growth so indolent that many months may pass before the patient is aware of the minimal, painless proptosis. CT scan will confirm the presence of an orbital
mass, but the image display is no different than the CT scan analysis of other small cell lymphomas of the orbit. The conjunctival tumor will be evident over a less number of months. Usually, the tumor will present as a painless, soft, reddish mass in one of the fornices of the conjunctiva. The literature has noted a delay in initial diagnosis because of the clinicians’ presumption that patients had some type of conjunctivitis. Other mistaken initial diagnoses include cicatricial entropion of unknown etiology, recurrent trichiasis, cicatricial pemphigoid, sebaceous carcinoma, and contact lens intolerance.
mass, but the image display is no different than the CT scan analysis of other small cell lymphomas of the orbit. The conjunctival tumor will be evident over a less number of months. Usually, the tumor will present as a painless, soft, reddish mass in one of the fornices of the conjunctiva. The literature has noted a delay in initial diagnosis because of the clinicians’ presumption that patients had some type of conjunctivitis. Other mistaken initial diagnoses include cicatricial entropion of unknown etiology, recurrent trichiasis, cicatricial pemphigoid, sebaceous carcinoma, and contact lens intolerance.
If, at the time of presentation, the lymphoma is primary in the orbit, several authors believe that bone marrow may also be affected. However, other authors do not support this assessment. Nevertheless, there is general accord that foci of tumor are more often associated with MALT tumor of the orbit when compared to tumor in the gastrointestinal tract. If, in the course of either conjunctival or orbital tumors, there is dissemination of tumor, it likely will spread to another extranodal site.
With light microscopy, the follicle components of the tumorous, polyclonal, benign lymphoid hyperplasia and MALT tumor are similar in appearance. Immunohistochemistry is helpful in the differentiation of the two lesions. The benign tumor will stain for different immunoglobulin light and heavy chains. The neoplastic cells of a MALT lymphoma are B cells that are negative for CD5, CD10, and cyclin D1.
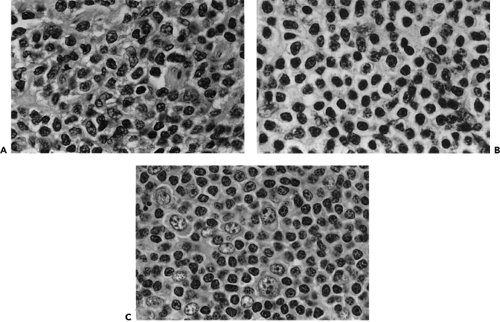 Figure 13.12 Morphologic spectrum of MALT lymphoma cells. A: Neoplastic marginal zone B cells with nuclei resembling follicle center centrocytes but with more abundant cytoplasm. B: The cells have abundant pale staining cytoplasm leading to a monocytoid appearance. C: The cells resemble small lymphocytes associated with larger transformed lymphoblasts. Magnification not stated. (From Jaffe ES, Harris NL, Stein H. World Health Classification of Tumors. Lyon: IARC Press; 2001:158. ) (See Color image.) |
In the histopathology of the MALT lymphoma, the cells infiltrate around the reactive B-cell follicles, external to a preserved follicle mantle, in a marginal zone distribution. These marginal zone B cells have small- to medium-sized, slightly irregular nuclei with moderately dispersed chromatin and inconspicuous nucleoli. The cells have relatively abundant, pale cytoplasm. Alternatively, the marginal zone cells may more closely resemble small lymphocytes (see Fig. 13.12A, B, and C) (Jaffe et al., 2001).
Hardman-Lea et al. (1994) have noted that, in the absence of systemic disease, a MALT lymphoma of conjunctival origin may be so innocuous that observation alone is sufficient. Two of their five patients were observed, with no local or systemic treatment. One patient was followed up for 30 months and the other for 12 months. Neither showed evidence of either local progression or
systemic dissemination. Thereby, the patients were spared some of the morbidity of radiotherapy. Likewise, a large, bulky MALT lymphoma that is either a cosmetic nuisance or interfering with ocular rotation can be surgically debulked. Radiotherapy may be reserved for the lymphomas that, over a passage of many months or several years, undergo a more malignant transformation of a large-cell clone. The MALT lymphoma of the orbit is said to be more aggressive than a conjunctival neoplasm. Low-dose radiotherapy is the initial, preferred treatment.
systemic dissemination. Thereby, the patients were spared some of the morbidity of radiotherapy. Likewise, a large, bulky MALT lymphoma that is either a cosmetic nuisance or interfering with ocular rotation can be surgically debulked. Radiotherapy may be reserved for the lymphomas that, over a passage of many months or several years, undergo a more malignant transformation of a large-cell clone. The MALT lymphoma of the orbit is said to be more aggressive than a conjunctival neoplasm. Low-dose radiotherapy is the initial, preferred treatment.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


