Fibrous and Connective Tissue Tumors and Proliferations
Fibrous tumors and tumor-like proliferations are a large and diverse group of entities, which differ greatly in behavior. Some are perfectly benign, remain localized, and usually do not recur even after simple excision. Others are poorly circumscribed, grow in an infiltrative manner, and tend to recur unless widely excised. Still others are frankly malignant, recur, and metastasize in a high percentage of cases. On the basis of age at incidence, Enzinger and Weiss (1995) divided them into four categories: (i) Benign fibrous proliferations, (ii) fibromatoses, (iii) fibrosarcomas, and (iv) fibrous proliferations of infancy and childhood.
Myofibroma and Myofibromatosis (Infantile Type)
Stout (1954) described a group of abnormal fibrous proliferations in patients younger than 16 years. To the tumors in this age-group he applied the term juvenile fibromatosis. Later, Chung and Enzinger (1981) reviewed the clinical and pathologic characteristics of 61 cases and to this tumor group they applied the term infantile myofibromatosis. “Infantile” replaced “juvenile” because 89% of their cohort occurred within the first 2 years of life. The prefix “myo-” was added because the microscopic features of the tumor cells were intermediate between fibroblasts and smooth muscle cells. The authors also recognized solitary and multicentric forms of the tumor. We prefer the terms “myofibroma” for the solitary tumor and “myofibromatosis” for the multicentric lesions because the form may vary according to the age at presentation (infancy, childhood, or adulthood). The suffix “-tosis” may be reserved for a discussion of a spectrum of multiple causes of the fibrous tumor.
Incidence
Only one publication in the literature attempts an analysis of the incidence of orbital involvement. In an extensive review of 340 orbital tumors in children, Kodsi et al. (1994) found only one case (incidence 0.3%) of orbital myofibroma. Our survey of 1,795 orbital tumors (see Table 3.3) includes four cases of fibromatosis (0.0022%).
In Table 5.1, we list the seven myofibromas reported in the literature since 1988. There are five males and two females. All but one patient were <4 years old at the time of presentation. Not included in the list is a case of desmoid fibromatosis reported by Maillard and Kountakis (1996). This case probably was associated with trauma. Myofibromas also occur in adults, usually in the lower eyelid, although the literature contains only a smattering of adult cases.
Clinical Features
Myofibroma is a gray, benign, nonencapsulated, locally invasive tumor that usually presents as a painless proptosis of several weeks’ to several months’ duration. If located in the inferior orbit, there is swelling of the overlying eyelid and a hard mass is palpable. Because the development is painless, there may be undue delay before the child is brought to the attention of the ophthalmologist. In such cases, there may be chemosis and redness of the eye and a look of alarm on the patient’s face.
Such patients may also have papilledema. All the neoplasms in Table 5.1 were solitary. Five of these seven patients had some degree of orbital bone erosion. In three of the five patients, erosion involved the bony interface between the orbit and the intracranial vault. The largest
tumor, measuring 5.2 × 5.4 cm (Stautz, 1991), entered the anterior cranial fossa through the lamina papyracea and optic foramen and entered the middle fossa through the greater wing of the sphenoid bone. All patients underwent some type of surgical intervention. Incisional biopsy was performed in three patients, and another three patients had total excision, piecemeal removal, or extirpation. The patient described by Campbell and Garrity (1991) underwent two surgical procedures (see Fig. 5.1). An initial incisional biopsy was performed. However, clinical progression of the tumor continued. Nine months later the tumor was excised through a lateral orbitotomy. Tumor regression occurred in all cases.
tumor, measuring 5.2 × 5.4 cm (Stautz, 1991), entered the anterior cranial fossa through the lamina papyracea and optic foramen and entered the middle fossa through the greater wing of the sphenoid bone. All patients underwent some type of surgical intervention. Incisional biopsy was performed in three patients, and another three patients had total excision, piecemeal removal, or extirpation. The patient described by Campbell and Garrity (1991) underwent two surgical procedures (see Fig. 5.1). An initial incisional biopsy was performed. However, clinical progression of the tumor continued. Nine months later the tumor was excised through a lateral orbitotomy. Tumor regression occurred in all cases.
Table 5.1 Myofibromas Reported Since 1988 | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Imaging Aspects
On computed tomography (CT) scan, because of the frequency of some degree of bone remodeling, the tumor probably appears circumscribed, with marginal sclerosis. Magnetic resonance imaging (MRI) shows isodense to high signal intensity on T1-weighted images, and T2-weighted and gadolinium-enhanced images demonstrate the high signal intensity of the tumor. The intensity of the enhancement may be useful in judging the degree of regression of the tumor in the follow-up period.
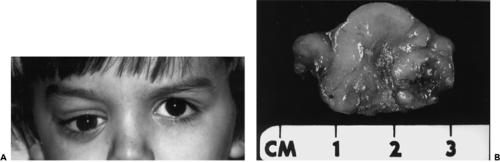 Figure 5.1 A 4-year-old boy with progressive forward and downward displacement of the right eye of 14 months’ duration owing to a hard palpable mass in the superoanterior orbit. B: Surgical specimen. The mass arose from the periorbita and measured 3.0 × 2.5 × 1.5 cm. |
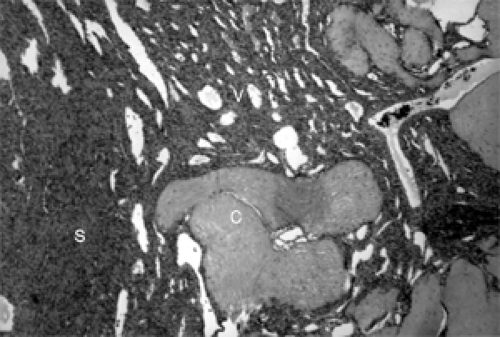 Figure 5.2 Cellularity of this myofibroma varies from area to area. Note solid spindle cells (S), vascular component (V), and dense collagen (C) (hematoxylin and eosin, original magnification × 160). |
Pathology
Histologically, sections from a myofibroma show marked variation of cellularity from area to area within a given tumor and from one tumor to another. This display is called a zoning pattern (see Figs. 5.2 to 5.4). The center of the tumor usually has some type of a vascular component, slit-like cavities, capillary channels, or a hemangiopericytoma-like pattern. This area is surrounded by an admixture of spindle, fusiform, and oval cells arranged in ribbons, fascicles, or bundles that vary in density from area to area. Areas of marked density may have deposits of dense collagen. Areas of less cellularity may show patches of loose fibromyxomatous stroma. Long-standing tumors show patches of necrosis and calcium deposition. Mitoses are either rare or absent.
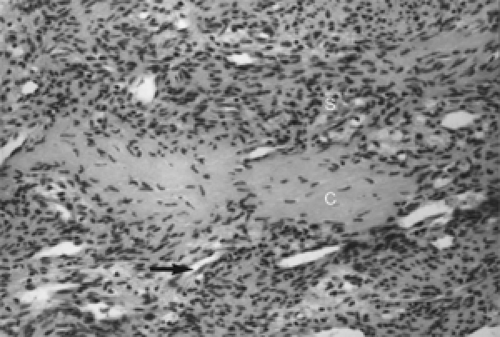 Figure 5.3 Spindle cells (S) of this myofibroma intertwine. Collagen (C) is surrounded by slit-like vascular channels (arrow) (hematoxylin and eosin, original magnification × 400). |
Immunoperoxidase stains are usually positive for vimentin (a general soft tissue marker) and actin (a marker for contractile protein), the latter a contractile protein. Some observers say these staining properties are variable. Staining is variable also for desmin (a soft tissue marker) but negative for S-100 protein (a marker of neural crest derivatives and other tissue) and leukocyte common antigen (a white blood cell marker).
Ever since Chung and Enzinger (1981) suggested that the tumor cells were intermediate between fibroblasts and muscle cells, the tumor’s pathogenesis (fibrous tissue or smooth muscle) has been debated. At present, this discussion is not germane to the clinical course and management of the tumor.
Management
Assuming the myofibroma is primary in the orbit, the ophthalmologist has several treatment options, depending on such factors as size and location of the tumor, severity of visual dysfunction, and the extent of bone erosion. If the mass is located in the anterior orbit and the displacement, motility, and vision of the eye are not seriously compromised, an effort may be made to excise the lesion.
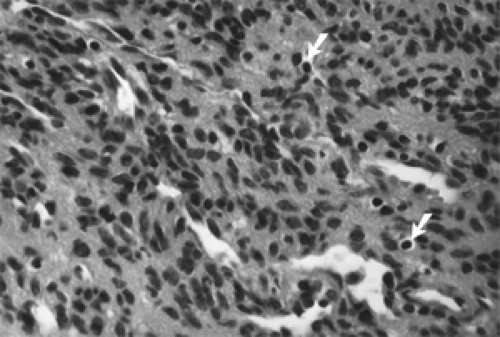 Figure 5.4 The central portion of this myofibroma shows spindle cells, sparse mononuclear cells (arrows), and slit-like vessels (hematoxylin and eosin, original magnification × 630). |
If the lesion is very large (because of either rapid progression or long duration), the vision and motility of the eye are severely restricted, and the patient is experiencing pain, piecemeal debulking is best. Some tumors of this size may also be undergoing some necrotic degeneration with the tumor in such a semigelatinous state as to be amenable to aspiration. Any tumor, regardless of size or location, which has papilledema or retinal disruption also should undergo debulking. Myofibromas that invade the bone, other than surface erosion, must be debulked, particularly if the sphenoid bone is involved.
Comment
To conclude this section, we should mention a 2½-year-old boy with a sino–orbital desmoid fibromatosis (aggressive juvenile fibromatosis) reported by Maillard and Kountakis (1996). Aggressive juvenile fibromatosis is a benign, poorly encapsulated, infiltrative, firm overgrowth of fibrous tissue, with a propensity to invade bone and soft tissue, occurring in children in the 2- to 8-year age range (and considered rare in the sinuses [Naidu et al., 1991, Thompson et al., 1991]). In these respects, it is similar to the preceding infantile myofibroma.
It differs from myofibroma, however, in several respects, namely, its aggressiveness, which belies its assumed benignity; its recurrence; and, if located near a vital area, its lethality. The authors believe their case was the sixth to be reported wherein the primary locus of the tumor was a paranasal sinus.
Within a month of the child falling and hitting his left cheek, the cheek began to bulge, pushing the left eye upward and deforming the left side of his face. Imaging studies showed a mass in the left antrum with extension into the ethmoid cells and erosion of both the orbital floor and the anterior maxillary wall.
The mass in the antrum was removed surgically within its pseudocapsule, but tumor extension into the orbit was dissected from the periorbita along the floor of the orbit. On re-exploration 3 months later, the sinus cavity had re-epithelialized. However, the infiltrating tumor was still present in the inferior orbit. This was vaporized with the carbon dioxide laser. Following a report that hormonal receptors have been identified in some cases of juvenile fibromatosis (Maddalozzo et al., 1993) adjuvant treatment consisted of a 2-month course of tamoxifen citrate (10 mg twice daily). MRI performed 2 years later showed no recurrence.
Nodular Fasciitis
Some years ago, Konwaler et al. (1955) described a circumscribed, subcutaneous tumor-like lesion that was
thought to be a distinct clinicopathologic entity. Although the lesion was alarmingly reminiscent of fibrosarcoma, the authors believed the entity was an inflammatory reaction. This proved a major step in the resolution of the confusion at that time regarding what constitutes a low-grade fibrosarcoma. Numerous clinicians and pathologists subsequently confirmed the basic premise of Konwaler et al. (1955) of an entity that, histologically, looked sarcomatous but, clinically, was benign. This feature was responsible for the term pseudosarcomatous fasciitis. In the early years, this term was the title of most descriptions of this lesion.
thought to be a distinct clinicopathologic entity. Although the lesion was alarmingly reminiscent of fibrosarcoma, the authors believed the entity was an inflammatory reaction. This proved a major step in the resolution of the confusion at that time regarding what constitutes a low-grade fibrosarcoma. Numerous clinicians and pathologists subsequently confirmed the basic premise of Konwaler et al. (1955) of an entity that, histologically, looked sarcomatous but, clinically, was benign. This feature was responsible for the term pseudosarcomatous fasciitis. In the early years, this term was the title of most descriptions of this lesion.
Incidence
This is a benign, self-limiting tumor composed of rapidly proliferating, immature fibroblasts and myofibroblasts in a cellular matrix with mitotic activity (atypical mitoses are rarely seen). The precise cause of the proliferation is unknown, but its histopathologic pattern supports a reactive process such as that seen with trauma, inconspicuous or otherwise.
It is one of the most common soft tissue lesions among the tumor and tumor-like lesions of fibrous tissues. Enzinger and Weiss (1995), at the Armed Forces Institute of Pathology (AFIP), reviewed >1,000 cases over a 20-year period. In the AFIP series, this tumor was most common in adults aged 20 to 40 years. In the series of 250 cases described by Shimizu et al. (1984), the age range was 1 to 76 years with a mean of 39 years. Sex incidence was nearly equal.
In the 1960s and 1970s, the tumor was extensively studied by physicians anxious to report cases of the recently described clinical entity. Font and Zimmerman (1966) reported ten cases involving the eye and ocular adnexa (eyelids, five cases; periorbital tissues, two cases; subconjunctival tissue overlying the scleral insertion of a rectus muscle, one case; eyebrow, one case; and corneoscleral limbus, one case). In the same year, Tolls et al. (1966) described a case originating in the Tenon capsule (fascia bulbi). Subsequent cases were reported in the eyelid (Levitt et al., 1969), fascia bulbi (Ferry and Sherman, 1974), eyebrow (Meacham, 1974), and episcleral mass (Holds et al., 1990). None of these patients had proptosis. However, one article (Levitt et al., 1969) did use the word “orbital” in the title. However, Perry et al. (1975) described a 34-year-old woman with proptosis due to a mass in the superior temporal quadrant of the left orbit. On excision, the mass was not attached to any structure, was encapsulated, and was very vascular. This does not fit the present description of nodular fasciitis.
The best-documented orbital case was reported by Kaw and Cuesta (1993) in a 10-year-old girl with a painless, rapidly growing mass causing medial deviation of her left eye. There was no history of trauma. On surgical exploration, a shiny white, circumscribed, but unencapsulated, firm mass, 2 cm in diameter, was removed from its attachment to the periorbita. Histologic study confirmed the diagnosis of nodular fasciitis.
Clinical Features
Most patients present with a rapidly growing mass or nodule of from 2 to 6 weeks’ duration. Around the ocular adnexa and orbit, the lesions are solitary and not associated with pain or tenderness unless the growth reaches a size that pushes on a nerve. The CT scan of the patient of Kaw and Cuesta (1993) simply documented a circumscribed mass. An MRI study was not performed.
Pathology
The histologic variability of this lesion was pointed out in the initial description by Konwaler et al. (1955). Its basic components are proliferating bundles of fibroblasts. In the early lesion, these spindled and ovoid, mitotically active fibroblasts proliferate in a helter-skelter fashion in a loose myxoid matrix containing many intercellular clefts. A rich capillary network is also present, and microscopically, the tumors may infiltrate the orbital muscle. As the tumor matures, the lesion becomes more cellular and compact, and the myxoid matrix is gradually replaced by collagen. The capillaries become less abundant, and some nuclei show prominent nucleoli. In the third stage, fibroblasts are more slender and compressed into whorls and interdigitating bundles similar to fibrous histiocytoma. Collagen deposition increases and the cell nuclei are more hyperchromatic. The content of mononuclear inflammatory cells varies considerably from specimen to specimen. These cellular variants represent a continuum of changes that correlate well with the duration of the lesion, the myxoid subtype having the shortest history and the fibrous subtype having the longest history.
Ultrastructural study reveals cells that seem to combine the properties of the fibroblast (abundant rough-surfaced endoplasmic reticulum and no basement membrane) and the smooth muscle cell (thin cytoplasmic actin filaments with fusiform densities, i.e., myofibroblasts) (Jakobiec and Jones, 1979).
Management
The orbital and adjacent adnexal growths are managed by simple excision.
Nasopharyngeal Angiofibroma
This long-known, benign rhinologic neoplasm may also be found in the orbital soft tissues. Several sizable, well-documented series of angiofibromas—120 cases by Neel et al. (1973) and 218 cases by Stern et al. (1986)—have been published wherein proptosis or displacement of the
eye occurred. Although the lesion is chiefly a secondary intruder into the orbital domain, this anatomic area may be an even rarer primary locus for the tumor.
eye occurred. Although the lesion is chiefly a secondary intruder into the orbital domain, this anatomic area may be an even rarer primary locus for the tumor.
Incidence
Neel et al. (1973) in their 120 patients noted orbital invasions in ten patients (8%). Stern et al. (1986) noted orbital involvement in a larger number of patients, in 26 (12%). In the interval since these reports, we have not found another large, well-documented collection of cases in the literature, which could be the basis for statistical analysis. Moreover, we believe that the frequency of orbital spread is much less because of factors not present in the 1970s and 1980s. First is the routine use of the CT scan at the time of the patients’ initial visits. The CT scan image shows the full anatomic extent of the tumor and ramifications outside the nasal passage. In turn, this imaging encourages earlier surgery and more complete excision of the primary tumor. Second, surgical technology has been refined.
All 120 patients in the series reported by Neel et al. (1973) were men. Some authors have stated nasopharyngeal tumors always occur in men. However, several reports have noted the tumor’s occurrence in women (Finerman, 1951; Osborn and Sokolovski, 1965; Conley et al., 1968; Peloquin et al., 1997). Rarely, patients with this angiofibroma also have familial adenomatous polyposis (Giardiello et al., 1993; Ferouz et al., 1995).
Clinical Features
The common presenting features are some degree of nasal obstruction, facial deformity, and repeated epistaxis. Examination shows a pale, red or reddish blue mass in the pterygopalatine fossa projecting into the roof of the posterolateral nasal cavity. Bulging of the adjacent cheek often is present because of the pressure erosion of underlying bone. A thorough bone imaging on CT scan shows the full extent of the tumor. Initially, some erosion of sphenoid bone occurs behind the sphenopalatine foramen (Lloyd et al., 2000). The deeper the tumor extension is into cancellous bone, the greater the likelihood some remnant of tumor may remain after surgical removal. Because of the high vascularity of the tumor, MRI shows signal voids and strong postcontrast enhancement. Because of its aggressive nature, the tumor rapidly expands into the retromaxillary area, the orbit, and the intracranial vault by a combination of passages through bony fissures and bone erosion. Orbital invasion is heralded by one or more of the following: Reduced vision, displacement of the eye, tearing, and ophthalmoplegia. Intracranial extension often occurs with or near the time of orbital invasion. Such patients have papilledema or optic atrophy.
The one case of angiofibroma in our tumor series was a 16-year-old boy with trouble breathing through the left side of his nose during the preceding 6 to 8 weeks. Two weeks before his referral, his referring physician biopsied a mass in the left nasal passage. The biopsy report indicated that the tumor was an atypical angiofibroma.
On admission, imaging studies revealed a large mass in the left posterior nasopharynx extending into the sphenoid, ethmoid, and maxillary sinuses. Through a lateral rhinotomy, the affected sinuses were exenterated, and the presenting surface of the tumor was treated by cryoablation.
Ten months later, angiography revealed extensive recurrence of tumor in the left nasopharynx, the floor of the middle intracranial fossa, and marked dilation of the ophthalmic vein. The left eye was proptosed 5 mm, and the anterior surface of the sclera was covered by dilated veins (see Fig. 5.5). The area was treated with cobalt 60, 24 Gy, over a 10-day period. At the last follow-up 3½ years later, the tumor had disappeared, and the eye looked almost normal.
Moschos et al. (1998) reported an angiofibroma they considered primary in the orbit. A 14-year-old boy presented with a mass in the left lacrimal sac area. The patient had had a previous surgery for dacryocystitis. Imaging procedures showed erosion of the lower nasal wall of the orbit with displacement of the eye temporally. The mass was totally removed. Pathologic study indicated a nasopharyngeal angiofibroma. We consider the lacrimal sac, anatomically, to be an adnexal structure that is outside the orbit. Therefore, this case is an angiofibroma secondarily invading the orbit. The case of a 15-month-old boy with an angiofibroma, anterior and medial to the lacrimal sac, described by Schick et al. (1998) is anatomically in the same area. The tumor was removed through an endonasal, microscopic–endoscopic approach. Finally, Weprin and Siemers (1991) noted a biopsy-proved juvenile nasopharyngeal angiofibroma first diagnosed in an 11-year-old child. The patient was followed up for 12 years but received no therapy. Total involution of the
lesion occurred. This involution supports the theory that this tumor may resolve with time.
lesion occurred. This involution supports the theory that this tumor may resolve with time.
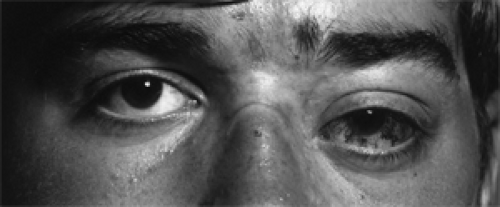 Figure 5.5 Recurrent nasopharyngeal angiofibroma. This 17-year-old man had total excision of a 10 × 7 × 4-cm multilobular mass 10 months previously involving nasopharynx, nasal cavity, sphenoid sinus, retromaxillary space, maxillary antrum, and cheek, all on the left side. Angiography showed an enormous recurrence involving the floor of the left middle intracranial fossa and the orbit, and obstruction of the ophthalmic veins. Note the dilated epibulbar vessels on the bulging, displaced left eye. The recurrent tumor was considered inoperable, but it responded to radiotherapy. The patient was living without further recurrence at follow-up 3 years after this photograph was taken. |
During the past several decades, the effect of sex hormones on the clinical course of angiofibromas has been debated. Two publications on the subject in the rhinologic literature of the 1990s should be noted. Using immunohistochemical methods, Gatalica (1998) examined eight nasopharyngeal angiofibromas for the expression of androgen receptors, estrogen receptors, and progesterone receptors. No estrogen receptors or progesterone receptors were found in the tumor components. Variable weak-positive androgen receptor immunoreactivity was found in a minority of endothelial and stromal cells, similar to normal turbinates. He concluded that the results argued against an important role for androgen receptors in the growth of these tumors. Hwang et al. (1998) performed immunocytochemical studies on 24 nasopharyngeal angiofibromas. Stromal and endothelial nuclear immunostaining was positive for androgen receptors in 18 (73%) of 24 cases, whereas 2 (8.3%) of 24 cases were positive with antibodies to progesterone receptors. None of the samples was positive with antibodies to estrogen receptors.
Pathology
The tumor is a profusion of thin-walled vascular channels of variable size and shape interspersed in a fibrous tissue stroma (see Fig. 5.6). The fibrous tissue consists of a complex proliferation of spindle and stellate cells in the more peripheral zones of the lesion, whereas a less cellular, predominantly collagenous matrix is usually present in the deeper, more central zone. The amount of the vascular component decreased as the extracellular collagenous fibers increased (Liang et al., 2000). In the more proliferative zone of the tumor, the endothelial cells of the vascular channels lie directly against the supporting stroma, without any intervening elastic fiber or smooth muscle cells (pericytes). This probably accounts for the profuse hemorrhage when the tumor is manipulated. In more collagenous areas, spindle cells may layer between vessel wall and stroma. If so, some of these cells probably stain positive to actin, indicative of smooth muscle properties.
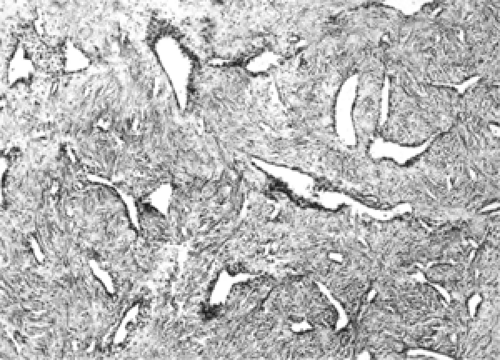 Figure 5.6 In this nasopharyngeal angiofibroma, a dense, cellular, spindle cell stroma supports a mixture of slit-like and irregular, angulated, thin-walled vascular channels. At this magnification, the tissue pattern resembles the spindle cell variant of hemangiopericytoma (hematoxylin and eosin, original magnification × 60). |
Management
The role of the ophthalmologist begins when and if the angiofibroma invades the orbit. It is likely that areas other than the orbit show extension of tumor on CT scan imaging. One, two, or more combinations of the following spaces may also be involved: Pterygopalatine fossa, maxillary sinus, sphenoid sinus, anterior intracranial fossa, maxillopterygoid fossa, infratemporal fossa, and middle intracranial fossa. Therefore, the multilocular angiofibroma may encroach on the boundaries of several surgical disciplines such as maxillofacial surgery, rhinologic surgery, plastic and reconstructive surgery, and neurosurgery, in addition to ophthalmology. Various surgical approaches recommended in the literature to remove tumor extensions include endoscopic, subtemporal, midfacial, lateral rhinotomy, lateral orbitotomy, and lateral preauricular temporal approach. For lesions around the cavernous sinus, microdissection has been recommended inasmuch as the tumor tends to push aside the structures rather than fuse with their fascial coverings. Before commencing these major surgical procedures, it is prudent to perform angiography and, if possible, to embolize the feeder vessels to the tumor.
Radiotherapy also is used as initial therapy but in recent years has been reserved for recurrent tumor after surgical removal. Reddy et al. (2001) analyzed 15 patients treated with radiotherapy between June 1995 and March 1996. Six patients received radiotherapy as the initial therapy, and the remaining nine had irradiation for recurrent tumor. All patients were boys between the ages of 11 and 14 years. Eight patients were followed up for 5 or more years, and seven were followed up for 10 or more years. The entire cohort was treated with continuous-course techniques with variable dosages ranging from 540 to 735 Gy. Thirteen patients had complete resolution of tumor within 1 to 39 months (median, 13 months). Two patients had tumor recurrence at 26 months and 28 months. Both these patients received irradiation as initial therapy. Cataract developed in three patients 5 to 10 years after irradiation. All three had tumor invasion of the orbit.
Kuppersmith et al. (2000) treated three patients with intensity-modulated radiotherapy. This technique embraces the concept of computer-controlled radiation deposition and computer-controlled normal tissue avoidance. One patient with an extensive tumor was treated initially with radiotherapy because the tumor was considered unresectable. The other two patients were treated for recurrent angiofibroma 11 months and 13 months after surgical
excision. The tumor in the first case eroded the base of the skull and extended into the pterygopalatine fossa and the area of the cavernous sinus. The tumor received a total dose of 45 Gy in 25 fractions. The mean dose to optic nerves was 28 Gy and to optic chiasm was 17 Gy. Imaging scans showed tumor regression at 15 months. The two patients with recurrent angiofibromas were treated with a total of 45 Gy and 35 Gy. Tumor resolution occurred at 40 months and 6 months, respectively. No further follow-up of the three cases was stated.
excision. The tumor in the first case eroded the base of the skull and extended into the pterygopalatine fossa and the area of the cavernous sinus. The tumor received a total dose of 45 Gy in 25 fractions. The mean dose to optic nerves was 28 Gy and to optic chiasm was 17 Gy. Imaging scans showed tumor regression at 15 months. The two patients with recurrent angiofibromas were treated with a total of 45 Gy and 35 Gy. Tumor resolution occurred at 40 months and 6 months, respectively. No further follow-up of the three cases was stated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


