Fibro-Osseous, Osseous, Cartilaginous, Vascular, and Inflammatory Tumors of Orbital Bone
This chapter covers an array of dysplastic, neoplastic, and odontogenic tumors affecting orbital bone. The fibro-osseous group of tumors is discussed first, inasmuch as it is a logical transition between the fibrous tissue tumors of the preceding chapter and the bony and cartilaginous tumors of this chapter.
Fibro-osseous Tumors
These lesions are an admixture of fibrous tissue and osteoid (immature bone that has not undergone calcification). The fibrous tissue usually is the predominant constituent with a content of osteoid that differs quantitatively and structurally from one tumor to another. This group includes fibrous dysplasia, fibro-osseous dysplasia, giant cell granuloma, aneurysmal bone cyst, and ossifying fibroma.
Fibrous Dysplasia
Fibrous dysplasia is a benign lesion characterized by the presence of fibrous connective tissue with a whorled pattern and trabeculae of immature nonlamellar bone. It is not considered a true neoplasm (Spraul et al., 1996). This lesion was first defined by Lichtenstein (1938), but its cause remains unknown. Jakobiec and Jones (1979) suggested the tumor was the result of an arrest of bone maturation at the woven bone stage. Most cases present in childhood or early adolescence. Initially, the disorder was predicted to arrest at puberty. Tanaka et al. (1993) described a boy with craniofacial fibrous dysplasia that showed marked involution at the end of puberty. Now, the tumor is known to progress well into adult life (Horgan et al., 1999).
Incidence
Our current 10-year survey (1988 to 1997) added six more primary orbital fibrous dysplasias (Table 3.2) to those tabulated in the previous editions of this book. The grand total of primary dysplasias observed over 50 years is 12, six males and six females, an incidence of 0.006% among 1,795 tumors (Table 3.3). The age range was 6 months to 63 years, with a median of 19 years. Bibby and McFadzean (1994) also reported a small series of 12 cases from the Leicester Royal Infirmary, London. There were five male and seven female patients. Age at diagnosis ranged from 5 to 45 years, with an average of 18 years. Most commonly involved were the frontal bone (eight patients) and the sphenoid bone (seven patients). Bilateral lesions occurred in three patients. They concluded that fibrous dysplasia is not a disease confined to adolescence but may continue into adulthood and sometimes middle age.
Clinical Features
The symptoms of this lesion depend on the anatomic site of the affected bone, the number of bones affected, the rate and duration of tumor growth, and the soft parts that are compressed, distorted, or displaced by the expanding
bone. The chief manifestation of this lesion is a unilateral, slowly progressive proptosis and displacement of the eye. Beyond this mass effect of the growing tumor on the eye, the presentation depends on the orbital location of the affected bone. If the tumor is well anterior in the frontal, ethmoid, or maxillary bones, the displacement of the eye in a direction opposite to involved bone may be equal to or greater in extent than the proptosis. Such patients may also have grotesque transmogrification of the upper face (see Fig. 6.1). Strangely, some such patients may retain good visual acuity and have little functional impairment of the eye other than a limitation of ocular motility in the field of the growing tumor and epiphora secondary to nasolacrimal duct compression.
bone. The chief manifestation of this lesion is a unilateral, slowly progressive proptosis and displacement of the eye. Beyond this mass effect of the growing tumor on the eye, the presentation depends on the orbital location of the affected bone. If the tumor is well anterior in the frontal, ethmoid, or maxillary bones, the displacement of the eye in a direction opposite to involved bone may be equal to or greater in extent than the proptosis. Such patients may also have grotesque transmogrification of the upper face (see Fig. 6.1). Strangely, some such patients may retain good visual acuity and have little functional impairment of the eye other than a limitation of ocular motility in the field of the growing tumor and epiphora secondary to nasolacrimal duct compression.
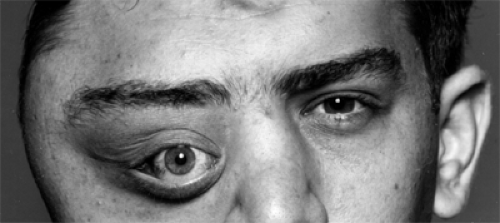 Figure 6.1 Fibrous dysplasia of the right temporal, frontal, malar, ethmoid, and sphenoid bones in a 19-year-old man with progressive proptosis, displacement of the right eye since the age of 8 years, and deformity of the right side of his face since the age of 10 years. The disease continued to progress over a period of 31 years. The patient underwent the last of seven surgical procedures, a craniotomy, at the age of 51 years to alleviate convulsions secondary to encroachment of the tumor on his brain. |
As the process extends posteriorly or if the tumor originates either in the posterior extent of the above-mentioned bones or the sphenoid bone, the proptosis increases in proportion to displacement of the eye, and visual function is more seriously affected. These patients more often have annoying, persistent headache or discomfort on the side of the lesion, particularly if there is compression of structures in the orbital apex, reflecting encroachment on cranial nerves III, IV, and VI. Usually, pain is not an important component of the presentation except when the tumor becomes so large that it competes with the displaced eyeball for limited orbital space. Stenosis of the bony optic foramen occurs from expansion of fibrous dysplasia of the sphenoid bone, the periorbita is stretched by a sudden intrusion into the orbital space by tumor from a paranasal sinus, or an unusual rapid growth of tumor. The latter is illustrated in the report by McCluskey et al. (1993). Over a period of only 4 months, a 41-year-old woman developed progressive diplopia, reduced visual acuity, pain in the left orbit, proptosis of 7 mm, and downward and inward displacement of the left eye associated with a large, tender mass in the fossa of the lacrimal gland. The bony mass already had eroded the upper lateral margin and frontal process of the zygomatic bone on the left side.
Other unusual patterns of presentation should be mentioned, particularly patients who experience extreme loss of vision in the affected eye: A 21-year-old woman with visual loss to light perception over 1 week responsive to high dose corticosteroid therapy (Osguthorpe and Gudeman, 1987), a 20-year-old woman with no light perception (Liakos et al., 1979); a 17-year-old woman who became blind in the affected eye 4 hours before admission (Ronner et al., 1982); a 13-year-old boy with counting finger vision (Melen et al., 1980); an 8-year-old boy in our series with no light perception; and a 3-year-old boy also with no light perception (Posnick et al., 1993). However, the nuances of the tumor’s presentation, symptoms, and course are about the same for most of the bone tumors in this chapter. In short, they are nonspecific in contributing to diagnosis of the specific type of bone tumor. All the signs and symptoms noted in the preceding text serve as a guide to the correct site for radiographic studies and for biopsies (Unni, 1996). Yuen et al. (2002) reported a third case in the literature of fibrous dysplasia associated with aneurysmal bone cyst.
Imaging Aspects
Computed tomography (CT) scan shows the size, shape, and extent of orbital bone involvement. Anatomic factors may affect the radiographic image of the tumor. In thinner bone, such as the orbital plate of maxillary, ethmoid, and frontal bone, the cortex expands more rapidly and to a greater degree than in thick bone. Therefore, thinner bones tend to show more lucency, cavitation, and compartmentalization of the dysplastic process.
Lucency equates with a lytic, cyst-like pattern of bone expansion. Thick bone (see Fig. 6.2), such as the sphenoid, tends to react in a more solid, sclerotic, and diffuse manner. Also, the dysplasia may transgress suture lines and affect multiple bones that rim the orbital space. In later stages, extension of tumor across the midline of the skull is not uncommon (see Fig. 6.3). Absence of a sharp border between cortical bone and the medullary constituents
of the tumor produces a homogeneous smudge on the radiograph. Nevertheless, differentiating intraosseous meningioma from fibrous dysplasia affecting the roof and apex of the bony orbit poses difficulties. Errors in diagnosis caused by excessive reliance on diagnostic imaging occurred on three occasions in a series of 25 patients reported by Hansen-Knarhoi and Poole (1994).
of the tumor produces a homogeneous smudge on the radiograph. Nevertheless, differentiating intraosseous meningioma from fibrous dysplasia affecting the roof and apex of the bony orbit poses difficulties. Errors in diagnosis caused by excessive reliance on diagnostic imaging occurred on three occasions in a series of 25 patients reported by Hansen-Knarhoi and Poole (1994).
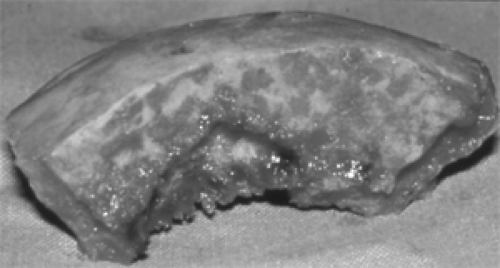 Figure 6.2 A 2-cm-thick piece of bone from the right frontal orbital plate of the man with craniofacial fibrous dysplasia pictured in Figure 6.3. The mottled areas on the cut surface of the bone (above) represent small, irregular, cyst-like spaces that contained a sanguinous fluid. The large cavity (lower left) was filled with tissue that suggested an aneurysmal bone cyst. (See Color image.) |
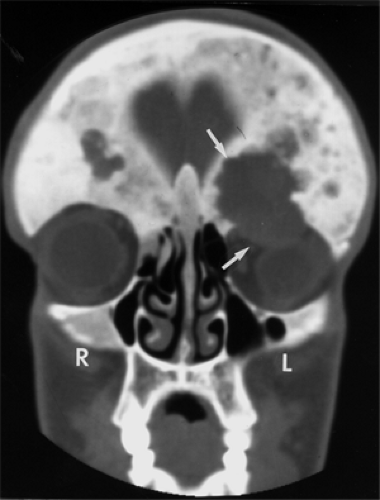 Figure 6.3 Coronal computed tomography scan with contrast enhancement from a patient with fibrous dysplasia shows mottled densities and lucent areas in both frontal bones, greater on the right than on the left. A large cavitary erosion of the left side of the orbital roof (upper arrow) by a soft tissue mass (lower arrow) extends inferiorly into the left orbital space, causing downward displacement of the eye. On surgical excision, the inferior soft tissue mass suggested an aneurysmal bone cyst. |
Casselman et al. (1993) reported magnetic resonance imaging (MRI) of five patients with biopsy-proved craniofacial fibrous dysplasia. According to this report,
Low to intermediate signal intensity was usually seen in the largest part of the lesion on both spin-echo sequences, but smaller regions of hyperintensity on T1– and T2-weighted images and intermediate signal intensity throughout a lesion on T1-weighted images are also seen. All lesions enhanced but only two became iso- or hyperintense compared to fat. High clinical and pathologic activity in three cases correlated with high signal intensity on both spin-echo sequences and with strong enhancement in two of the three. The presence of large veins or sinusoids on pathologic examination did not correlate with the enhancement pattern.
Pathology
Fibrous dysplasia mirrors the early stage of intramembranous ossification of the bones of the skull in which osteoid is formed by mesenchymal-derived spindle cells (fibro-blasts). This is illustrated in Figure 6.4, which shows spicules of woven bone interspersed in a stroma of uniform, benign spindle cells. There is no nuclear atypia and mitoses are rare. With polarized light, the woven bone shows an uneven birefringence. If the affected bone had developed normally, osteoblasts would then have appeared at the periphery of the bone elements and would have converted the woven bone into the lamellar form. Multinucleated giant cells are occasionally present. With the passage of time, the stroma may develop acellular myxoid-like zones of degeneration.
Management
When the third edition of this text was written, more vigorous surgical approaches evolved to replace the conservative efforts to remove fibrous dysplasia of orbital bone, which was the norm of prior decades. The aim during the 1980s was to remove all dysplastic bone by staged surgery, to alleviate blindness, if necessary, by decompression of the optic nerve, and to reconstruct or revise the contour of the bony defect.
Surgical management was further refined in the 1990s to remove all dysplastic bone and reconstruct the orbit in
one surgery and to prevent optic nerve dysfunction by early decompression, rather than delaying surgery until vision was already impaired to some degree (Mahapatra et al., 2003).
one surgery and to prevent optic nerve dysfunction by early decompression, rather than delaying surgery until vision was already impaired to some degree (Mahapatra et al., 2003).
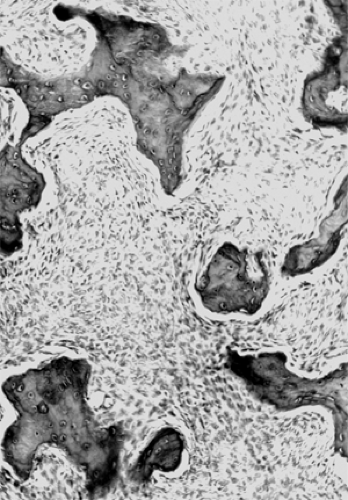 Figure 6.4 Fibrous dysplasia. Irregular contoured islands of woven bone are interspersed in a spindle cell stroma. Unlike ossifying fibroma, there are no osteoblasts rimming the osseous spaces (original magnification × 125). |
Perhaps the largest series of cases relevant to refinements in the surgical management of orbital fibrous dysplasia is that of Yavuzer et al. (2000). One of the coauthors, I. T. Jackson, MD, has had extensive experience in this field over the time of several editions of this text. Yavuzer et al. (2000) treated 32 cases of fronto-orbital fibrous dysplasia during the past 20 years and assessed the results of treatment. The treatment consisted of radical resection of the fibro-osseous tissue, decompression of the optic nerve canal, and reconstruction of the fronto-orbital areas either with noninvolved bone graft or with dysplastic bone that was contoured down or heated in the sterilizer. During follow-up, two tumors recurred, but repeat optic nerve decompression was not required. Overall, the aesthetic results were satisfactory. Patient ages ranged from 2 to 51 years, with a mean age of 16.8 years.
Ricalde and Horswell (2001) also commented on their experience with the surgical management of six patients with fronto-orbital surgery. Their patients ranged in age from 7 to 28 years. Surgery generally involved extensive tumor excision and immediate orbital reconstruction with autogenous bone grafts. Two patients also had reconstructive surgery with resected and treated autogenous bone, which was then immediately replanted using rigid fixation. Three patients had intracranial microsurgical optic canal decompression. All patients received postoperative corticosteroids. Five patients experienced partial relief from their sensory and visual disturbances. One patient who underwent two-wall optic canal decompression had visual loss. They recommended early surgery to avoid the hazards of late-stage decompression.
Papay et al. (1995) addressed the surgical decompression of the bony optic canal in cranial base fibrous dysplasia. Seven coauthors were included, thereby indicating their support for a multispecialty approach for surgery of the anterior cranial base. They described five patients without measurable loss of visual acuity who underwent superior decompression of the optic canal in the course of radical excision of dysplastic bone. However, imaging displays showed early signs of visual afferent defects, venous congestion of the superior ophthalmic vein (color Doppler display), or a combination of these. No ophthalmic morbidity was encountered during the mean follow-up of 18 months.
This more vigorous approach and earlier management of major craniofacial dysplasias have come about chiefly in the past decade. The publications that support these concepts cite one or more of an array of reasons for altering the conservative management stance of former years: To correct deformity of the face and periorbital region, to alleviate headache and pain attendant to expansion of the craniofacial bones, to relieve proptosis of the eye, to forestall further visual loss where such a functional deficit is already present, to alleviate sudden intralesional hemorrhage, and to eradicate malignant transformation of the dysplasia where such malignancy is either proved or suspected. Less direct are the reasons that emphasize the prevention aspects of aggressive and early management such as avoiding the delayed effects of progression of the dysplasia and preventing complications while awaiting spontaneous arrest of the disease. The last-named reasons may be more wishful thinking than factual confrontation with the disease.
In general, the most predictable result of these major surgical ventures is subjective more than objective improvement in the bony disfigurement of the patient’s skull or face. Less often is the patient unequivocally pleased with the postoperative position of the eye, particularly if reconstruction of the floor of the orbit was either attempted or necessary. A new, disconcerting, long-lasting diplopia often occurs postoperatively, chiefly because of an unavoidable shift in mechanics of the inferior and superior oblique muscles. Blepharoptosis of some degree and variable duration is almost always a complication of radical surgery of the frontal bone. Even so, the most worrisome problem confronting both patient and surgeon is the ultimate effect on the visual function of the eye. Will the patients with failing vision be better or worse after surgical decompression of the optic nerve? The odds appear to be about even either way. Will the patient without preoperative visual disability lose vision as the result of a surgery involving dysplasia of the sphenoid bone or base of the skull? Here the results are “all or none.” Postoperatively, either the visual function is unscathed or the patient is blind. No data are available to answer this question because the cases with favorable outcomes are reported more often than cases that are visual disasters.
The question of the “preventive” purpose of these surgeries should also be addressed. Will early surgery delay progression of the disease? Probably so, although a major drawback in all the reports by enthusiasts for the surgery is the lack of long-term follow-up data. Will the surgery prevent complications while the patient waits for spontaneous arrest of the dysplasia? Probably not!
Our experience with six patients observed over 10 to 32 years may give some clue to the answer to the last question.
Case 1
A 19-year-old man had fibrous dysplasia of five cranial bones on the right side. Although visual acuity of the right eye was normal, early pallor of the right optic disk was present. This was worrisome for future visual loss because of threatened encroachment of dysplastic bone on the right optic canal. This factor, plus the marked disfigurement of the right side of his face and displacement of the right eye (Fig. 6.1), prompted the decision to proceed with craniofacial surgery. Over the next 10 months, five staged surgical procedures were performed to alleviate the
situation. The disfigurement was greatly improved, and the vision remained stable until, 6 years later, proptosis reappeared, pallor of the optic disk increased, and vision dropped to 20/200, all in the right eye. In another 6 years, by the age of 31 years, pallor of the left optic disk also was noted, and a right upper quadrant hemianopsia was present in the visual fields, indicating an optic tract involvement. At the age of 39 years, convulsive seizures commenced, hearing loss on the right side occurred, and the hard palate was involved by the advancing dysplastic process, in addition to further progress in all prior signs and symptoms. At the last follow-up, at the age of 51 years, the patient had undergone another craniotomy to relieve a left hemiparesis secondary to encroachment of the dysplasia on the brain.
situation. The disfigurement was greatly improved, and the vision remained stable until, 6 years later, proptosis reappeared, pallor of the optic disk increased, and vision dropped to 20/200, all in the right eye. In another 6 years, by the age of 31 years, pallor of the left optic disk also was noted, and a right upper quadrant hemianopsia was present in the visual fields, indicating an optic tract involvement. At the age of 39 years, convulsive seizures commenced, hearing loss on the right side occurred, and the hard palate was involved by the advancing dysplastic process, in addition to further progress in all prior signs and symptoms. At the last follow-up, at the age of 51 years, the patient had undergone another craniotomy to relieve a left hemiparesis secondary to encroachment of the dysplasia on the brain.
Case 2
A 10-year-old girl with fibrous dysplasia involving three bones of the skull underwent a right craniofacial surgical procedure because of persistent headache and right-sided craniofacial disfigurement of 1 year’s duration. Subsequently, the patient was free of pain for 4 years and then underwent another craniotomy for recurrence of headache. A pain-free interval of 7 years elapsed before another craniotomy, including a section of a division of the trigeminal nerve, which was necessary for recurrent pain. A fourth craniotomy for recurrent pain was performed at the age of 24 years. The right eye was blind after this surgery. By the age of 29 years, the recurrent head pain was treated with repeated nerve blocks and surgical section of peripheral nerves. The patient was also medicated for temporal lobe epilepsy. Imaging of the skull at this time indicated extension of the dysplasia across the intracranial midline.
Case 3
This 8-year-old boy was blind in the right eye from fibrous dysplasia of the sphenoid bone. A debulking procedure was performed, including unroofing of the right optic canal. Postoperatively, the right eye remained blind. Skull films at the age of 11 years showed recurrent dysplasia in the lesser wing of the right sphenoid bone. At the age of 14 years, the patient was subjectively and objectively stable. However, by the age of 29 years, radiography showed extension of the dysplastic process into the right frontal bone, the right middle intracranial fossa, and the left sphenoid bone. Vision in the left eye was not affected.
All these three patients showed definite progression of their disease into early and late adult life. Two of the three were seriously handicapped by their fibrous dysplasia. The third case seems definitely at risk for eventual intracranial neurologic defects.
Case 4
A 23-year-old woman with 11-mm proptosis of the left eye and fibrous dysplasia of three cranial bones underwent a craniofacial excision of tumor chiefly to relieve disfigurement. The latter was deemed successful until drainage appeared in the operative site 19 months later, following what was considered minor trauma to the left side of her forehead. This was treated with hot packs and antibiotics followed by remission. Over the next 16 years, the patient continued to receive local therapy intermittently because of recurrence of drainage from the surgical site. There had been no further surgical procedures in this interval, and the patient had not experienced any visual difficulty, but information concerning the imaging status of her dysplasia was incomplete.
Case 5
A fibrous dysplasia localized to the orbital plate of the left frontal bone with erosion into the intracranial space in a 15-year-old girl was excised through an anterior orbitotomy. A 2-mm left blepharoptosis was a permanent sequel to the surgery. Ten years later, the patient’s ocular and orbital statuses were stable, and there was no obvious progression of the dysplasia. However, follow-up radiography was not done. Even so, she is the only patient among those described herein who had 10 years without some complication of either progressive disease or the surgical procedure.
Case 6
The sixth patient, a 14-year-old girl, underwent excision of a fibrous dysplasia of the ethmoid bone with orbital extension in April 1987. However, follow-up on this patient was only 14 months, too short an interval to objectively assess a disease as resilient as fibrous dysplasia.
Several conclusions seem warranted from the above discussion.
A long follow-up period is necessary before judging the arrest or progression of fibrous dysplasia. Well-documented cases of spontaneous arrest of the orbital disease seem to be lacking. A minority of patients with dysplasia limited to either the frontal or maxillary bones seem most amenable to total excision and possible cure.
Most of the patients with dysplasia of two or more cranial bones probably have slowly progressive disease for an indefinite period in their life span. Such patients may ultimately have serious neurologic complications, particularly if the sphenoid bone is initially affected. The extension of the disease in such patients probably is related to the unavoidable, limited excision of the lesion that is possible at the base of the skull. At present, there are no standard guidelines or criteria for the management of this distressing and potentially disabling disease, except that a debulking procedure may be considered for some patients with headache or pain.
Finally, there is some hope in the medical management of patients with extensive involvement of bone
proximal to vital orbital structures where surgical intervention poses a high risk for functional loss, particularly in children. Yavuzer et al. (2000) have used a bisphosphonate, pamidronate, which seems to arrest invasion of fibrous dysplasia into adjacent normal bone by limiting osteoclastic activity, which causes normal bone resorption. The drug is only a temporizing measure as long as the treatment regimen is maintained. The drug is administered as an intravenous infusion on an outpatient basis. Three doses of 60 mg or 1 mg per kg in children is given at intervals of 1 day to 1 week. This regimen is repeated every 6 months, or more frequently, depending on the patient’s clinical picture.
Ossifying Fibroma
Kempson (1966) originally described this neoplasm. Although considered benign, it is also a lesion of aggressive growth. Tumors that originate in the paranasal sinuses or at the base of the skull usually affect the orbit secondarily. The tumor is closely related to fibrous dysplasia and may actually be a variant of fibrous dysplasia. The numerous spherules of bone that are scattered throughout the stroma of the lesion prompted Margo et al. (1985) to add the qualifying term “psammomatoid” to distinguish this tumor from other fibro-osseous tumors.
Incidence
Margo et al. (1985) selected from the file of the Armed Forces Institute of Pathology 21 patients with ossifying fibroma of orbital bone. The average age of their group at the time of initial diagnosis was 17.8 years, with a range of 4 months to 52 years. The sex incidence was equal. Many clinical details of this cohort, including the laterality of the lesion, were not stated.
Table 6.1 Ossifying Fibroma of Orbital Bones: Reports of Individual Cases | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
In the third edition of this text, we culled 14 cases of orbital ossifying fibroma reported by nine authors between 1964 and 1988. The mean age of these patients was 15.5 years, with a median of 10 years. The male-to-female ratio was 9:5. This indicates the prevalence of the tumor in the first 2 decades of life. Two cases of bilateral orbital involvement were included in the survey, a 22-year-old woman (Scott et al., 1971) and an 8-year-old boy (Margo et al., 1986) (see Table 6.1).
Five additional cases of pathologically proved orbital ossifying fibroma have been reported since 1988: A 7-year-old boy (Palma et al., 1988); a 16-year-old boy (Perri et al., 1996); a 12-year-old boy (Takaya et al., 1993); a 29-year-old man (Nakagawa et al., 1995); and a 6-year-old boy (Fakadej and Boynton, 1996).
Clinical Features
The chief manifestation of orbital encroachment is a slowly progressive, painless proptosis and displacement of one eye associated with diplopia. More serious functional and ophthalmic deficits, such as visual loss, optic atrophy, and paralytic strabismus, usually do not occur until the lesion extends into the orbital apex or sphenoid sinus. Such signs and symptoms are also common portents of tumor growth. The tumor’s slow evolution was confirmed by the study of Margo et al. (1986). In their group of 21 patients, the average duration of symptoms before initial diagnosis was 4.6 years. Only in four patients was the duration <12 months. This presenting mode is essentially the same as that of a patient in the same age-group with fibrous dysplasia.
Encroachment of the orbital space comes from tumorous expansion of the orbital plates of the frontal, ethmoid, and maxillary bones, roughly in that order of frequency.
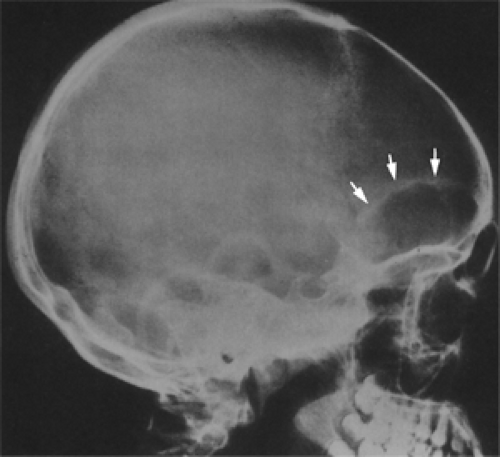 Figure 6.5 Psammomatoid ossifying fibroma. Lateral skull radiograph shows a radiolucent lesion expanding the roof of the orbit (arrows) into the anterior cranial fossa. The lesion is encompassed by very thin sclerotic bone. (From Margo CE, Shields JA. Diagnosis of fibro-osseous bone lesions [Letter]. Ophthalmology. 1989;96:569–570 , with permission.) |
Imaging Aspects
Radiography (see Fig. 6.5) and CT scan are the imaging mainstays of fibro-osseous tumors involving the orbit, particularly if more than one bone is affected. The tumor is a destructive process that tends to distort and expand the affected bone but retains a thin, somewhat sclerotic shell of peripheral bone. The lesion is round or ovoid in contour, and on plain film radiography, it appears radiolucent in a solid bone and radiopaque when adjacent to an air-filled sinus. In the early stage, the lesion is confined to one bone and the margins are well defined. With the passage of time, the lesion expands to additional bones, and in its late stages, it may show productive changes that seldom compensate for the early osteolysis.
CT scan shows the patchy-appearing, nonhomogeneous matrix with its mixture of radiodense and radiolucent material. Also, there may be a radiolucent zone between the tumor’s matrix and the surrounding unaffected bone, which tends to emphasize the tumor’s circumscription. Because of its slow, painless evolution, the lesion may reach considerable size. In 12 of the 21 cases studied by Margo et al. (1985), the average diameter of seven lesions affecting the frontal bone was 5.3 cm and that of five lesions of the ethmoid plate was 5.5 cm. On MRI scans, ossifying fibromas appear heterogeneous and usually show intermediate signal on T1-weighted and hypointense signal on T2-weighted MRI. Contrast enhancement is moderate on gadolinium-enhanced T1-weighted MRI scans (Wenig et al., 1998).
Pathology
Grossly, the tumor has a smooth surface, is covered by a thin shell of sclerotic bone, and, in the orbital plates, tends to be multicystic. It consists of both fibrous and osseous elements, with the former predominating. The fibrous stroma is highly cellular, vascular, and well endowed with fibroblasts with plump nuclei, particularly in tumors of short duration. In older lesions, the fibroblasts are more compressed, with flattened nuclei. Some mitotic activity is present in all lesions.
The most diagnostic features, however, are the size, configuration, and makeup of the numerous spherules of lamellar bone scattered throughout the stroma. Most of these bone elements are ovoid and are surrounded by a delicate pink-staining layer of osteoid, which, in turn, is rimmed by osteoblasts. The rim of osteoblasts is prominent in contrast to their inconspicuousness in typical areas of fibrous dysplasia.
Other histopathologic components of the lesion, such as intercellular collagen, trabeculae of woven bone here and there, and occasional giant cells, are minor in quantity and consistency. Two variants of ossifying fibroma deserve mention. One is the cementifying fibroma that likely originates from cells along the periodontal area of the teeth in the upper jaw. It is also a destructive tumor that, by progression, may secondarily invade the orbit. The calcified material in this lesion is cementum rather than bone. Rarely, these tumors seem to arise in orbital bone, as the tumor did in the 12-year-old boy reported by Takaya et al. (1993). These lesions probably originate from cells capable of producing cementum as well as bone and fibrous tissue. This admixture of tissue could be called a cemento-ossifying fibroma.
The other variant is the psammomatoid ossifying fibroma that typically arises in the sinonasal passages and invades the orbit through the ethmoid sinus and medial orbital wall. Their distinctive component is the mineralized or calcified ossicles, of variable size, scattered throughout the bony trabeculae and adjacent cellular stroma (see Fig. 6.6).
Surgical Management
When the manuscript for this section was completed in 1989 for the third edition of this text, we indicated it was likely that, in the future, excision of the tumor would be accomplished in most cases at the time of the initial surgery. This would supersede the multistage surgical procedure used before 1989.
Now, almost all authors writing on the subject of surgical management of ossifying fibroma strongly advocate complete removal of the tumor at the time of initial surgery. If multiple bones are involved, they recommend a multidisciplinary approach to tumor removal by surgeons from the various surgical disciplines accustomed to working in the anterior portions of the skull. They also believe that
an open (combined craniofacial transfrontal procedure) surgical approach assures complete removal of the tumor compared with endoscopic procedures often used with paranasal sinus disease. The latter is often confounded by postoperative imaging that shows residual tumor. In such cases, these aggressive tumors do recur. Several reports have reviewed surgical options, surgical details, and follow-up data on three or more patients (Blitzer et al., 1989; Lawton et al., 1997; Hartstein et al., 1998).
an open (combined craniofacial transfrontal procedure) surgical approach assures complete removal of the tumor compared with endoscopic procedures often used with paranasal sinus disease. The latter is often confounded by postoperative imaging that shows residual tumor. In such cases, these aggressive tumors do recur. Several reports have reviewed surgical options, surgical details, and follow-up data on three or more patients (Blitzer et al., 1989; Lawton et al., 1997; Hartstein et al., 1998).
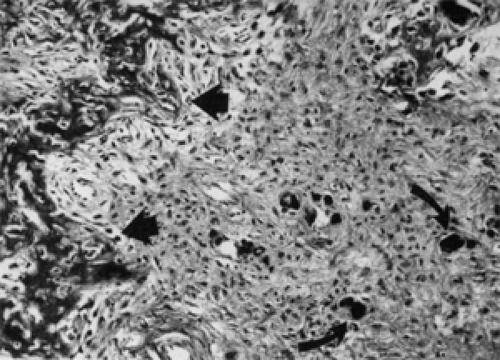 Figure 6.6 Psammomatoid ossifying fibroma shows immature woven bone (straight arrows) in a transition area between the small, round ossicle pattern of bone formation and normal bone, which is located toward the left. Several small, round ossicles (curved arrows) are also frequent in this transition area (hematoxylin and eosin, original magnification × 160). (From Margo CE, Shields JA. Diagnosis of fibro-osseous bone lesions [Letter]. Ophthalmology. 1989;96:569–570 , with permission.) |
Fibro-osseous Dysplasia
Unni (1996) favored this designation for a group of nonneoplastic lesions that consist of a large component of tissue elements of fibrous and mesenchymal origin (fibroblasts and spindle cells), both resorption and formation of bone, and giant cells. Three of these lesions are unique, inasmuch as they have essentially the same histopathology but differing clinical features. These are giant cell reparative granuloma, cherubism, and brown tumor of hyperparathyroidism.
Pathology
Grossly, these lesions are soft, spongy, multicystic, and friable with a reddish brown color and gritty consistency. In orbital bone, they are intraosseous and tend to expand the bone. When the fragile exterior is ruptured, the tumor may contain a black, viscous fluid consistent with old blood. The dominant cellular component is the benign fibroblast with benign multinucleated giant cells interspersed throughout the stroma (see Fig. 6.7). There is no cellular atypia. Mitotic figures are present in the fibroblasts but not in the giant cells.
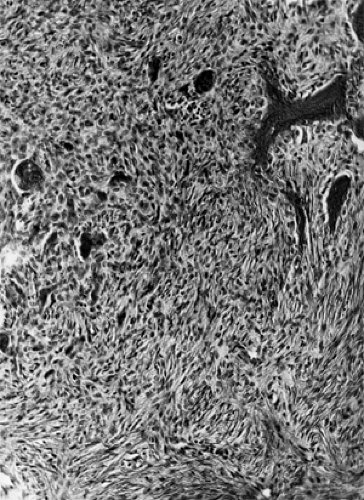 Figure 6.7 In this section from the mandible of a patient with fibro-osseous dysplasia, a few benign giant cells are present, but the histologic pattern is dominated by fibroblastic cells. Osseous metaplasia, almost always present, is seen at the upper right. (From Unni KK. Dahlin’s bone tumors: General aspects and data on 11,087 cases, 5th ed. Chapter 28, Philadelphia, PA: Lippincott-Raven; 1996 , with permission.) |
If hemorrhage has occurred, giant cells and epithelial histiocytes tend to cluster around the hemorrhagic zone. Almost all specimens show new bone formation as well as bone destruction. Southgate et al. (1998) showed the multinucleated giant cells in surgical specimens from a case of cherubism to be osteoclasts “since they synthesised tartrate- resistant acid phosphatase, expressed the vitronectin receptor, and resorbed bone.” The cause of this cellular response is not known except in the brown tumor of hyperparathyroidism. Here, increased levels of calcium and parathyroid hormone in the peripheral blood cause increased osteolysis.
Giant Cell Reparative Granuloma
When Jaffe (1953) described this lesion, he noted a tendency of the giant cells to surround foci of hemorrhage. He assumed the hemorrhage was secondary to trauma and the giant cells represented some type of healing or reparative process. Subsequently, the association of this tumor with a history of trauma has not been consistently reported, and the word “reparative” has been abandoned by many authors. The present trend is to simply designate the tumor as “giant cell granuloma.” However, the lesion
is still considered to be some type of “reactive” process, but the cause remains elusive.
is still considered to be some type of “reactive” process, but the cause remains elusive.
In the skull, the granuloma predominantly affects the mandible, maxilla, and temporal bone, roughly in that order of frequency. Over time, it has also been observed in the frontal, ethmoid, and sphenoid bones, with concomitant orbital encroachment. The orbital lesions are locally aggressive and, along with proptosis, displacement, and some limitation of ocular rotation, are associated with pain, particularly the intraosseous tumors. The expansion of the thin plate of orbital bone stretches the periorbita, thereby causing the pain. In addition, a certain amount of bone destruction produces an additional inflammatory response. By contrast, the presentation of fibrous dysplasia is usually painless unless it is exceptionally aggressive and rapid in growth. The histopathology of this tumor is illustrated in Figure 6.8.
Those cases are either primary in orbital bone or show encroachment of the orbit from a locus of tumor in an adjoining sinus (Sood et al., 1967; Friedberg et al., 1969; Hoopes et al., 1981; Rhea and Weber, 1983; Case Records of the Massachusetts General Hospital, 1984; Chapman, 1984; Sebag et al., 1985; Spraul et al., 1997; Hyver et al., 1998; Mercado et al., 1999). The age range of the 15 patients reported is 5 to 83 years, with an average of 24.2 years and a median of 11.5 years at the time of diagnosis. There were 13 males and 2 females in this group.
CT scan shows an orbital mass with a heterogeneous internal structure and adjacent bone erosion. MRI shows a circumscribed intraosseous cystic mass with internal signals consistent with blood and fluid levels (Mercado et al., 1999).
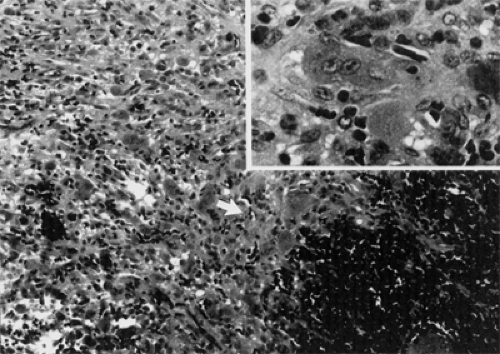 Figure 6.8 An infiltrate of epithelioid histiocytes (arrow) and giant cells surrounds a focus of hemorrhage at the lower right (hematoxylin and eosin, original magnification × 50). Inset: Histiocytes are shown at a higher magnification (hematoxylin and eosin, original magnification × 250). (From Unni KK. Dahlin’s bone tumors: General aspects and data on 11,087 cases, 5th ed. Chapter 28, Philadelphia, PA: Lippincott-Raven; 1996 , with permission.) |
Most surgeons involved in the surgical removal of these lesions, in and around the orbit, are members of a multidisciplinary surgical team comprising neurosurgeons, head and neck surgeons, otorhinolaryngologists, ophthalmologists, and plastic surgeons. The object of surgery is complete removal of the lesion, and this is usually possible.
Cherubism
In 1933, William Jones, a Canadian radiologist, reported a familial intraosseous multilocular cystic disease of the jaws in three siblings of the same family, characterized by “full round cheeks” and “upward cast of the eyes” that gave the children a “cherubic appearance.” Jones coined the term “cherubism,” because he thought the children resembled the cherubs frequently painted in Renaissance art.
The histopathology of this lesion is the same as giant cell reparative granuloma and brown tumor of hyperparathyroidism. At birth, the shape of the head and face is normal. At the age of 2 or 3 years, hard, bilateral, symmetric, painless masses usually appear around the angle of the mandible and spread to the ascending rami and body of the mandible. Next, the maxillae are involved, but the extent of the disorder varies from minor lesions to massive involvement of both jaws. The teeth are irregularly placed and may be missing. About 200 cases of cherubism have been reported in the literature, but orbital involvement is infrequent.
Orbital involvement occurs when and if the upper portions of the maxillary bones are involved. This causes bulging of the inferior orbital plate which, in turn, pushes the eyes superiorly. This displacement results in varying degrees of diplopia, tearing, and proptosis. If there is massive involvement of maxillary bones with long-standing upward expansion of the inferior orbital floor, some degree of visual loss occurs from compression of the optic nerve. This is rare, however, either because the disorder spontaneously self-arrests or because the bony deformity is surgically removed before this complication. The disorder usually subsides at puberty, with the affected bone reverting to some semblance of its normal contour. However, there are some exceptions.
One unusual exception was the 27-year-old woman described by Colombo et al. (2001). The patient had a slowly progressive, bilateral superonasal globe displacement and temporal orbital masses of 6 years’ duration. Although there was a history of cherubism, her cheeks and jaws appeared normal, and CT scans demonstrated multicystic bony lesions arising from the orbital floors bilaterally. The nature of the masses (cherubism) was confirmed histopathologically. The authors concluded that orbital involvement may develop beyond puberty, after stabilization or regression of the lesions in the jaws. Seven other patients have been reported with orbital involvement: Three by Jones (1933), two by Hawes (1989), one by Carroll and Sullivan (2001), and one by Schultze-Mosgau et al. (2003).
CT scan shows multilocular areas with intervening solid tissue involving the mandible and maxilla with extension along the floor of the orbit.
Anderson and McClendon (1962) established the genetic basis for the disorder. The gene locus was localized to chromosome 4p16.3. Cherubism is a familial disease transmitted in an autosomal dominant manner, with 80% to 100% penetrance and variable expressivity (Peters, 1979).
The management of these cases is a mixture of conservatism and aggressive surgical debulking. In the early stages, if one or two teeth are malformed or partially resorbed, these can be removed. However, if there is major disruption of bone in the tooth-bearing areas, surgical debulking of the osseous lesion is performed. Upward displacement of the eyes caused by expansion of the maxillary bones along the floor of orbital bone can be followed up by CT scans so long as only the anterior orbit is involved. If imaging methods show extension toward the posterior orbit, the anterior maxillary wall can be partly removed for access to the orbit, and the orbital floor debulked subperiostially.
Hyperparathyroidism (Brown Tumor)
von Recklinghausen (1891) described the changes in bone that were subsequently called generalized osteitis fibrosa cystica. Twenty years passed before the demineralization of bone, so characteristic of the disease, was associated with enlargement of the parathyroid gland. However, the question of whether the bone disorder caused the hyperplasia of the parathyroid, or vice versa, remained in dispute. Amazingly, another 20 years passed before this question was settled, and hyperparathyroidism was identified as the triggering mechanism (Jaffe, 1933).
In the primary form, such as a neoplasm or hyperplasia of the parathyroid gland, the metabolism of bone goes askew, resulting in diffuse demineralization, elevated levels of calcium in the urine and blood, and a diminished serum phosphorus level. In secondary hyperparathyroidism, such as occurs in chronic renal failure, these assay values in serum and urine are reversed. The parathyroid gland responds by secreting parathyroid hormone in larger quantities.
In some cases, whether the cause is primary or secondary, the bone disorder is focal, such as in the orbit, rather than generalized in type. In the focal type, bone resorption is followed by cyst formation and an influx of reactive vascularized fibrous tissue. Hemorrhage and an influx of giant cells ensue. The sum of this process is a radiographically visible tumor of bone that resembles giant cell reparative granuloma and cherubism.
When incised or resected, these lesions are brown. But “brown tumor” is not an appropriate designation because the other two histopathologically similar tumors, giant cell reparative granuloma and cherubism, are also brown. Furthermore, the histopathologic features of the brown tumor are not pathognomonic. Diagnosis is best established by the elevated serum levels of calcium, alkaline phosphatase, or parathyroid hormone and an increased amount of urinary calcium.
Parrish and O’Day (1986) published a review of the literature pertaining to “brown tumor of the orbit.” They summarized what little is known about clinical presentation, course, and frequency of the tumor in the orbital bone. They analyzed 12 cases in the literature from 1953 through 1986. There were four males and eight females, ranging in age from 7 to 71 years (average, 30.4 years; median, 24 years). In five of the twelve, the lesion was of the primary type, six were associated with glomerulonephritis, and in one case the type was not stated. All patients presented with either some “swelling” of the eye, proptosis, or a palpable orbital mass. Reduced vision and pain were present in two cases. All cases underwent surgical excision of the tumor.
Two cases were added to this list and detailed in the third edition of this text. One was the 69-year-old woman described by Rootman (1988); the other was our patient, a 27-year-old woman listed as having “brown tumor” in Figure 6.9. The lytic lesion of the patient described by Rootman (1988) was well localized to the roof of one orbit and was completely resected. Histopathologic evaluation and subsequent serum assays of calcium, phosphorus, and parathyroid hormone led to the discovery of a parathyroid adenoma, which was removed.
Several years before presentation in our patient, a tumor had been removed from her right mandible at another institution. This tumor was thought to be a giant cell reparative granuloma. Plain film radiography demonstrated an expanding lesion of the right frontal bone. This osteopathy involved the orbital roof and greater wing of the sphenoid bone. Also, it extended intracranially (see Fig. 6.9A). The tumor was resected through a frontal craniotomy. The tumor also extended into the orbit through a lytic area in the orbital roof (Fig. 6.9B). Subsequent blood serum assays of calcium and phosphorus levels confirmed the diagnosis. One month later, an adenoma of the parathyroid gland was removed. The patient was followed up for 7 years without the development of any additional osteopathy. In retrospect, the jaw and orbital bone lesions probably represented multifocal lesions of hyperparathyroidism.
Finally, Levine et al. (1991) described a 27-year-old woman with hyperparathyroidism secondary to chronic renal failure. She had been receiving hemodialysis treatment for several years. Over the past decade or so, hemodialysis has increasingly been used for treatment of chronic renal failure. Therefore, the number of patients with secondary hyperparathyroidism has increased.
Aneurysmal Bone Cyst
This is a benign lesion of bone. The name aneurysmal bone cyst was first applied by Jaffe and Lichtenstein (1942) to differentiate this tumor from a solitary bone cyst. They noted the course of the tumor in two patients.
Subsequently, there was much debate about the pathogenesis of the lesion.
Subsequently, there was much debate about the pathogenesis of the lesion.
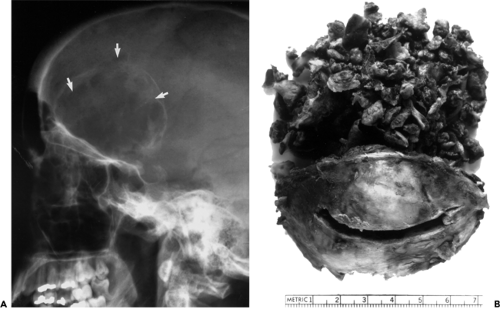 Figure 6.9 A: Sagittal radiograph showing an expansile lesion of the frontal bone with involvement of the orbital roof and the greater wing of the sphenoid bone of a 27-year-old woman. Note the large balloon-like, bosselated intracranial extension (arrowheads). B: En bloc resection shows gross specimen, measuring approximately 7 × 6 × 5-cm, attached to the orbital roof. The large upper portion of the lesion was extradural. The tufted configuration of the upper periphery corresponds to the bosselation noted in (A). |
Now, it is not considered to be either a cyst or a neoplasm. Instead, it probably is a pathophysiologic process that evolves from incomplete resolution of a tumor-associated vascular malformation. In approximately one third of cases, the preexisting lesion can be identified. Most common is the giant cell tumor. Other precursor tumors include osteoblastoma, chondroblastoma, fibrous dysplasia, nonossifying fibroma, solitary bone cyst, fibrous histiocytoma, eosinophilic granuloma, osteosarcoma, and angioma of bone. If no preexisting tumor is known, trauma (hematocele) should be considered. Aneurysmal bone cysts have been described in almost all areas of the skeleton except possibly the bones of the middle ear. Its favorite haunts are the long bones, vertebrae, ribs, and sacrum. We are chiefly concerned with its occurrence in and around the orbit (see Fig. 6.10).
Frequency
In the 1994 edition of this text, we referenced 17 single cases of unilateral orbital aneurysmal bone cyst in the literature from 1968 through 1990 (Fite et al., 1968; Offret et al., 1971; Komorn, 1972; Delorit and Summers, 1975; Powell and Glaser, 1975; Jakobiec and Jones, 1976; O’Gorman and Kirkham, 1976; Yee et al., 1977; Flament and Forest, 1979; Iraci et al., 1980; Ronner and Jones, 1983; Sanerkin et al., 1983; Klepach et al., 1984; Johnson et al., 1988; Rootman, 1988; Carmichael et al., 1989; Hunter et al., 1990).
Seven single cases of aneurysmal bone cysts were reported between 1993 and 2002 (Bealer et al., 1993; Patel et al., 1993; Dailey et al., 1994; Lucarelli et al., 1995;
Hino et al., 1998; Menon et al., 1999; Senol et al., 2002). Our 50-year survey list includes three cases, which brings the total number of cases to 27.
Hino et al., 1998; Menon et al., 1999; Senol et al., 2002). Our 50-year survey list includes three cases, which brings the total number of cases to 27.
 Figure 6.10 Displacement of the left eye caused by an aneurysmal bone cyst in a 5-year-old girl. The cyst occupied the upper third of the orbital space. |
These patients ranged in age from 11 months to 43 years (average, 13.6 years; median, 11 years). There were 17 females and 10 males. The orbital bone affected was frontal in 15, sphenoid in 6, ethmoid in 4, malar in 2, and maxilla in 1. The salient features of this statistical analysis are the 11:6 female-to-male sex ratio, a median age of 11 years for the presentation of the tumor, and the frequency of frontal bone involvement.
Clinical Features
In most anatomic sites, the principal sign of this tumor is an expansion of bone with subsequent stretching of the covering periosteum. The latter accounts for the principal symptom, pain of increasing severity. However, around the orbit, the bones are not thick enough to support a fusiform expansion of extended duration except for the body of the sphenoid, the base of the zygomatic arch, and the brow area of the frontal bone. Even in these sites, continuous pain of increasing degree is seldom encountered except in patients with involvement of the sphenoid bone. In such patients, ipsilateral headache is a frequent initial symptom of a growing cyst.
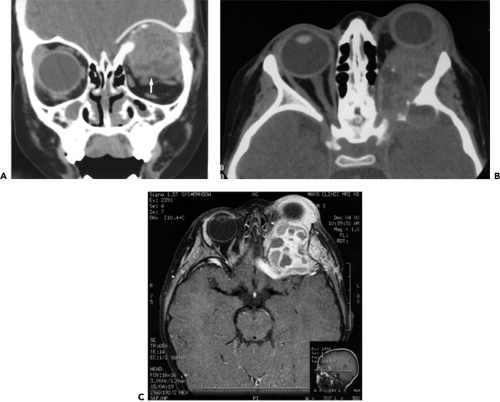 Figure 6.11 Imaging aspects from a 3-year-old boy. A: Computed tomography scan (coronal view) of massive tumor with irregular densities in the superoposterior orbit located in the peripheral orbital space between periorbita (arrow) and orbital bone. B: Axial view showing tumor invasion of the intracranial vault through erosion of the sphenoid bone. C: Magnetic resonance imaging axial view showing a “soap bubble” configuration. |
Nevertheless, in approximately 40% of the lesions in all orbital sites, pain may be the principal reason for seeking consultation. In these situations, the pain is usually of sudden onset and has been present for only a week or two. Such symptoms probably represent a sudden increase in the size of the lesion and stretching of the periorbita secondary to recent hemorrhage. In reality, the hemorrhage or sudden
expansion of tumor is superimposed on an otherwise insidiously growing tumor of unknown duration.
expansion of tumor is superimposed on an otherwise insidiously growing tumor of unknown duration.
In the approximately 60% of patients without pain or headache, proptosis is the chief presenting symptom owing to the bulk effect of the nonosseous part of the lesion usurping the orbital space. Such is the situation in most patients with frontal bone involvement. These patients show few signs or symptoms other than nonaxial proptosis and some mechanical limitation of gaze in the direction of the expanding mass. Occasionally, these patients may have an episode of ecchymosis of the upper eyelid or transient blepharoptosis if the cyst is located well forward along the orbital roof.
Patients with more serious orbital symptoms, such as optic disk edema, third and sixth nerve palsies, visual decrease, and an afferent pupillary defect, are those with sphenoid bone involvement or an intracranial extension of tumor. Nevertheless, suspicion of an orbital aneurysmal bone cyst is usually based on one of the imaging methods rather than the presenting clinical features.
Imaging Aspects
CT scan is particularly useful in defining the size and extent of lesions located in areas where the bony areas are complex, for example, the base of the skull. Fluid levels are common and may be seen on both CT scan and MRI. MRI seems superior for imaging the hemorrhagic content of the tumor because of its high-intensity signal on T1-weighted sequence. Also, MRI shows the internal trabeculations and the variable densities within the pseudocompartments of the tumor.
Kransdorf and Sweet (1995) studied the natural history through four evolving radiologic stages: Initial appearance, growth, stabilization, and healing. In skeletal bone, the initial stage is characterized by a well-defined area of osteolysis with discrete elevation of the periosteum. This is followed by a growth phase, in which the lesion grows rapidly with progressive “destruction” of bone and development of the characteristic “blown-out” radiologic appearance. The growth phase is succeeded by a period of stabilization, in which the characteristic “soap bubble appearance” develops, as a result of maturation of the bony shell. Final healing results in progressive calcification and ossification, with the lesion transformed into a dense bony mass. Figure 6.11 illustrates some of these imaging features.
Pathology
The consistency of the nonosseous portion of the tumor has been described as friable, fleshy, fibrous, or granular. This particular characteristic may depend on the age of the mass. The blood-filled spaces visually lack a lining of endothelium. All spaces may be separated by thin, delicate septa of loose connective tissue or by thicker compact bands of fibroblasts containing osteoid. Throughout the tumor are variable mixtures of hemosiderin-filled macrophages, benign giant cells, and lymphocytes. Some mitotic figures may be present (see Fig. 6.12). Calcification may be seen in both the fibrous stroma and chondroid material. The tumor on occasion may be a secondary phenomenon to chondroblastoma or fibrous dysplasia.
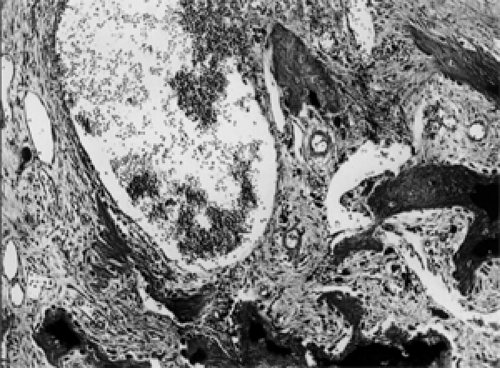 Figure 6.12 Blood-filled spaces are separated by fibrous septa containing bone spicules (original magnification × 110). |
Treatment and Prognosis
An orbital aneurysmal bone cyst usually requires only one surgery for satisfactory resolution, provided the surgical intervention is definitive rather than simply exploratory. In the orbit, the thin shell of bone typically covering the advancing form of the cyst in other anatomic sites may be absent. In the latter situation, there is only a covering of periorbita. The soft portions of the tumor are usually easily removed from the periorbita. The underlying defect in bone is curetted lightly, but persistently, to remove all loose, porous bone. Bleeding usually stops when this is accomplished. In cases where the orbital roof has been breached by intracranial extension of tumor, the soft tissue is wiped or sucked away from the covering dura. If this is accomplished, it is rare that a second operation is required. Cure of the lesion also has been reported after simple biopsy or incomplete removal. We do not believe radiotherapy is necessary for orbital lesions. If the lesion recurs, it usually appears within 2 years.
Osseous Tumors
Osteoma
This section discusses lesions with a predominantly osseous composition. Although the soft tissue component of these tumors exercises some supportive role, it is only a minor portion of the overall bony structure of the growth. It is the osseous feature of these lesions that determines the clinical manifestations and course of the tumor and poses a challenge in their management. The osteoma is the most common of this family of tumors with
orbital orientation, excluding those that have occurred after overzealous irradiation around the cranium.
orbital orientation, excluding those that have occurred after overzealous irradiation around the cranium.
This lesion is peculiar to the membranous bones of the face and skull. A favorite locale for its orbital presentation is the juncture of the bones of the ethmoidal and frontal sinuses in the anterosuperonasal orbit.
Osteoma has been classified into three types—ivory, mature, and fibrous—depending on the histopathologic proportion of bone to supporting fibrous stroma and the assumed maturity of the tumor. The fibrous type is considered the most immature and may be a continuum of or a link with the aforementioned ossifying fibroma. Apart from some minor variation in age range and rate of growth, the clinical course of the several histologic types is the same. Therefore, this subtyping is of little clinical importance.
In spite of much-published rhetoric and conjecture, the pathogenesis of osteoma is not known.
Incidence
The 50-year Mayo Clinic series includes 12 orbital cases, approximately 0.6% of our total series of pathologically proved lesions. This is a lower frequency than in other large study series (300 or more total orbital tumors) in the literature. Nine patients were men. Although the sex incidence of osteomas of the paranasal sinuses has been widely studied in a large number of patients (male-to-female ratio of 1.6 among 567 cases reported after 1939) (Mansour et al., 1999), that of primary orbital osteomas has usually not been included as a separate ratio.
The same omission in reference to the age range of patients with orbital involvement also occurs. Because osteoma of the orbit is slow growing, the tumor may remain asymptomatic for many years before presentation, thereby concealing the age at onset. Therefore, Grove (1978) noted an age range of 12 to 51 years based on the time of onset of symptoms rather than the time of diagnosis. The age range of our group of 12 patients based on the age at onset was 11 to 71 years (mean, 33.4 years; median, 32.5 years).
In our group of 12 patients, 4 had primary orbital osteomas and 8 had secondary extension of tumor from an adjacent sinus. Actually, primary orbital osteomas are very rare. Andrews (referenced by Cushing, 1927) found eight orbital osteomas among the records of 429,989 cases seen at three major ophthalmologic institutes in New York City.
The paranasal sinuses are, by far, the most frequent source of osteomas of the skull. The most frequent sinuses of origin are frontal (50%), ethmoid (40%), maxillary (6.2%), and sphenoid (3.6%) (Mansour et al., 1999). However, in terms of orbital extension, the ethmoid sinus is the most frequent source of osteoma.
A number of single cases of orbital osteoma have been recorded in the literature since the third edition of this text. All were osteomas of one or more paranasal sinus that secondarily extended into the orbit: A 32-year-old man (Biedner et al., 1988); a 14-year-old boy (Gillman et al., 1997); a 53-year-old woman (Gossios et al., 1999); a 15-year-old boy (Sires et al., 1999); a 66-year-old woman (Mansour et al., 1999); and a 13-year-old girl (Kim et al., 2000).
Chief among the oddities surrounding osteomas are the several references in the third edition linking osteoma of the jaw or skull with Gardner syndrome (Gardner, 1951). This is an autosomal dominant, familial polyposis of the large bowel, with epidermal and sebaceous cysts of the subcutaneous tissues. Also, the orbital osteoma may be a presenting manifestation (proptosis) of the disorder (Tandon et al., 1988). The importance of this association is that a malignant transformation of the intestinal polyposis may occur in nearly 40% of cases (Smith and Calcaterra, 1989; McNab, 1998).
Clinical Features
The anatomic site of orbital encroachment of the growing, expanding mass is the chief determinant in the ultimate pattern of presentation. In this respect, osteomas closely mimic the signs and symptoms of a mucocele in the same location; these were described for mucocele in Chapter 4. Osteomas originating in a paranasal sinus may remain asymptomatic until the sinus is filled with mature bone. Pain becomes a problem when the tumor pushes through the covering plate of bone and extends into the orbit. Osteomas arising in the intracranial vault have more room for unfettered growth and reach “giant” size before invading the orbit. Osteomas from a frontal sinus have one of the optional patterns of proptosis and downward displacement of the eye, the ethmoidal osteoma produces a more lateral shift of the eye, and the rare osteoma rising in the sphenoid sinus produces a modified orbital apex syndrome.
The rock-like consistency of an osteoma arising from the intracavitary surface of the bony orbital plate brings the patient with a primary orbital osteoma to the physician. It is the mechanical obstructive effect on ocular rotation that causes trouble. Biedner et al. (1988) reported a 32-year-old man who was unable to elevate the left eye when adducted (Brown superior oblique tendon sheath syndrome) because of an osteoma that had pushed the trochlea forward. Other cases of monocular, gaze-evoked, or transient loss of vision due to orbital osteoma were reported by Wilkes et al. (1979), Miller et al. (1977), and Gillman et al. (1997). In a patient described by Rootman (1988), an orbital osteoma was responsible for two episodes of subluxation of one eye in a 12-month period. In the 17-year-old boy described by Cecire et al. (1988), the osteoma was so unyielding that it eroded both the ethmoid plate and the periorbita, allowing air to enter the orbit when the patient blew his nose. In the case described by Mansour et al. (1999), cellulitis occurred when an osteoma of the ethmoid sinus broke into the orbit.
Imaging Aspects
CT scan is the procedure of choice to outline the full extent of the osteoma. The hyperdense mass probably shows some expansion of the sinus wall of origin (see Fig. 6.13). Some
tumors contain areas of spongy (cancellous) bone. If so, the signal intensity of CT scan is less in these areas compared with the surrounding compact bone (Bilaniuk et al., 1992). The tumor is not associated with bone destruction.
tumors contain areas of spongy (cancellous) bone. If so, the signal intensity of CT scan is less in these areas compared with the surrounding compact bone (Bilaniuk et al., 1992). The tumor is not associated with bone destruction.
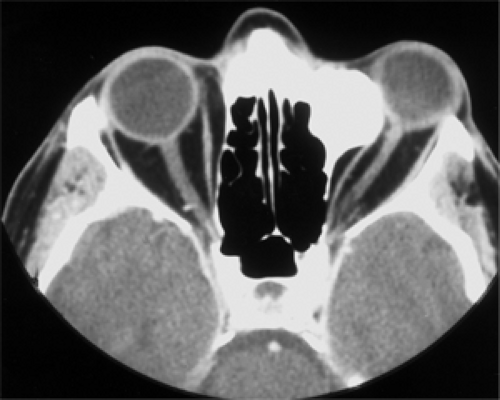 Figure 6.13 The computed tomography scan shows a 3 × 2.5 × 1-cm, hyperdense, smooth-surfaced osteoma along the superomedial, left orbital wall. The mass was completely resected. There was no recurrence 27 months later. |
On MRI, the signal void of an osteoma is difficult to distinguish from the signal void of air within the sinuses. However, the periorbital and dural coverings of the orbital plates show enhancement with MRI.
Pathology
The orbital osteoma, simply put, is an overgrowth of bone in the wrong place. Its structure is either cancellous or compact, lamellar bone or a mixture of both, depending somewhat on the site of origin or the maturity of the tumor. In the cancellous type, the trabeculae of bone are thinner, and the loosely arranged fibrovascular matrix is more abundant compared with the compact, eburnated bone (see Fig. 6.14). In the latter, the ivory-like bone is dense and thick, and the scanty fibrovascular component is compressed into tiny spaces, the haversian canals. Tumors arising from the orbital face of the bony wall have a covering of periorbita, whereas tumors of sinus origin also have part of their periphery covered with respiratory epithelium (see Fig. 6.15). Grossly, the compact subtype has an ivory-white, glistening surface, but the cancellous subtype may have a slightly pink hue. Some of the tumors are round and smooth, and others have a knobby, irregular contour (see Fig. 6.16).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


