Purpose
To describe the clinical findings in 3 patients with Duane syndrome and 3 different chromosomal duplications that may indicate the location of genes involved in the pathogenesis of this ocular motility disorder.
Design
Observational case series.
Methods
setting: Clinical practice. patient or study population: Three patients with Duane syndrome and chromosomal duplications from the clinical practice of 1 of the authors. observation procedures: Chart review and retrieval of clinical data and results of pertinent clinical tests, in this case chromosomal studies. main outcome measure: Reporting of details of clinical findings and duplicated chromosomal regions.
Results
Two patients had unilateral type I Duane syndrome and 1 had bilateral type I Duane syndrome. Two had cognitive delay, and all 3 had other systemic abnormalities, including a variety of congenital malformations. The chromosomal abnormalities that were detected using microarray analysis were 2q13(RP11-20G1,RP11-461N11) × 3, 10q24.2q26.3(101,532,585-135,284, 169) × 3, 20q13.12(44,796,613-44,945, 818) × 3, and 22q11.1q11.22(RP11-701M12, RP11-71G19) × 3.
Conclusions
Patients with Duane syndrome and associated congenital malformations or developmental delay should be evaluated for the presence of underlying chromosomal duplications. The regions of chromosomes 2, 10, and 22 that we report may harbor genes involved in the pathogenesis of Duane syndrome.
Duane syndrome refers to a spectrum of congenital ocular motility abnormalities that result from dysinnervation of the extraocular muscles. Most cases have anomalous innervation of the lateral rectus by a branch of the third rather than the sixth nerve and lead to type 1 Duane syndrome, characterized by lack of abduction beyond midline, normal adduction, and globe retraction with co-contraction of the medial and lateral rectus muscles in attempted adduction. Types 2 and 3 are also recognized, in which there are other ocular motility defects. There are also a multitude of less common and unusual forms of Duane syndrome with combinations of anomalous motility patterns that defy the normal laws of ocular motility. The genetics of Duane syndrome are complex, and although 2 genes have been associated with particular complex forms of Duane syndrome, the etiology in most patients remains unclear. A number of cases have been reported in association with chromosomal rearrangements. We present 3 patients who have Duane syndrome and hitherto unreported associated chromosomal duplications.
Case Reports
The first patient was referred because of type I Duane syndrome of the left eye ( Table ). She was born by cesarean section at 31 weeks gestation because of maternal pre-eclampsia and fetal distress. She weighed 2 pounds and was 14 inches in length at birth, and had an uneventful stay in the neonatal intensive care unit. She gained weight slowly and was followed by a geneticist and a pediatric endocrinologist because of failure to thrive and meet developmental milestones. The diagnosis of Duane syndrome was made at the age of 10 months. Patching and glasses were recommended, but she did not cooperate with either. Aside from Duane syndrome, short stature, and a hemangioma over her right scapula, physical exam was unremarkable. In light of a history of inadequate caloric intake and a family history of constitutional short stature on the patient’s maternal side, it was originally believed that the patient was likely a normal child who was slow to catch up developmentally from a premature birth.
| Patient | Chromosomal Abnormality | Motility Disorder | Other Clinical Findings |
|---|---|---|---|
| 1 | 2q13(RP11-20G1,RP11-461N11) × 3 | Left type I Duane syndrome |
|
| 2 | 10q24.2q26.3(101,532,585-135,284, 169) × 3, 20q13.12(44,796,613-44,945, 818) × 3 | Left type I Duane syndrome |
|
| 3 | 22q11.1q11.22(RP11-701M12, RP11-71G19) × 3 |
|
|
Microarray analysis and fluorescence in situ hybridization (FISH) was performed at the age of 5 years because growth retardation had not improved. The analysis was conducted using the SignatureChip 4.0 microarray (Signature Genomic Laboratories, LLC, Spokane, Washington, USA). A duplication of chromosome 2 at 2q13(RP11-20G1,RP11-461N11) × 3 was found. The nearest proximal clone that was not duplicated was RP11-468G5, and the nearest distal clone that was not duplicated was RP11-265B3. RP11-265B3 overlaps with RP11-461N11, so the distal boundary of the duplication is well defined. RP11-468G5, however, is 13.5 Mb upstream of RP11-20G1, so the proximal boundary is poorly defined. This duplication was not confirmed by FISH, based upon criteria that at least 70% of cells must show a duplication pattern to be reported. Therefore the duplication may be below the resolution of the FISH assay.
On initial presentation to our pediatric eye service, it was discovered that the patient’s father had been diagnosed with amblyopia, but the cause was unknown. The patient’s vision was 20/30 OD and 20/80 OS. She had hypermetropic astigmatism in both eyes. She was unable to abduct her left eye, and this eye drifted upward in attempted abduction. Her palpebral fissures narrowed in adduction, but adduction was 80% of normal. Her pupillary, external, slit-lamp, and fundus examinations were otherwise unremarkable.
The second patient was referred to our clinic at the age of 19 years because of left type I Duane syndrome and a partial trisomy of chromosome 10. He was born full-term with a birth weight of 6 pounds 8 ounces, and with several congenital anomalies. His head examination is significant for an irregular hair growth pattern; prominent brow; thick eyebrows and eyelashes; triangular, low-set, posteriorly rotated ears with simple helices; narrow nasal bridge; pointed nasal tip; thick columella; bifid uvula; dental crowding; and a small chin ( Figure 1 ). He has narrow shoulders, gynecomastia, and an asymmetrical posterior thorax with kyphosis. Musculoskeletal examination reveals limited motion of the shoulders, elbows, wrists, and fingers; hyperextended left wrist with ulnar deviation; and broad thumbs. His left leg is longer than the right and he has an inability to straighten the left leg. The right knee is larger than the left and he has an inability to fully extend the right knee. He has limited ankle flexion, his feet are turned outward, and the great toes are angulated. Spasticity is present in all 4 extremities, and the patient has a wide-based gait. The patient also has global developmental delay with mental retardation, bowel and bladder incontinence, and a hypoplastic right kidney.
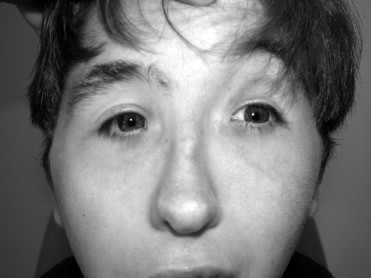
This patient underwent chromosomal staining and FISH analysis shortly after birth, demonstrating the presence of extra material on the end of chromosome 2p, but the origin of this material could not be identified at the time. His parents were found to have normal chromosomal staining patterns. At the age of 19, microarray analysis and repeat FISH was conducted by the ChildLab Cytogenetics Laboratory of Nationwide Children’s Hospital in Columbus, Ohio, USA using the SignatureSelect OS 105K v1.1 microarray (Signature Genomic Laboratories, LLC). This microarray identified trisomy of a 33.75-Mb region of chromosome 10 (10q24.2q26.3(101,532,585-135,284,169) × 3), a region containing over 300 genes. A trisomy of 20q13.12 was also discovered, though this region contains only the 3′ portion of 1 gene, SLC2A10 . FISH demonstrated that the extra chromosome 10 material was translocated to chromosome 2p25.3, with no loss of material at 2p. The extra copy of 20q13.12 was adjacent to the normal copy and was thought unlikely to be of clinical significance.
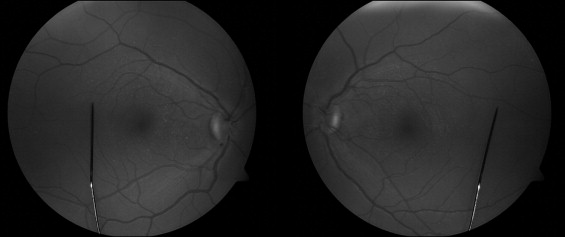
In addition to Duane syndrome, the patient’s ophthalmic history was significant for accommodative esotropia treated with glasses and patching beginning at 4 years of age. He had a remote history of head tilting, but this was attributed to torticollis and was corrected with cervical fusion surgery at the age of 6 years. On presentation to our clinic, the patient’s corrected visual acuity was 20/20 in the right eye and 20/40 in the left eye, demonstrating amblyopia. Abduction of the left eye was limited to 10 degrees, while adduction was limited to 50 degrees and elevation was limited to 40 degrees. The right eye had normal motility. In addition, the patient had blepharophimosis, thick long lashes, iris transillumination, and an anomalous optic nerve that appeared smaller than normal in diameter, with fine macular and extramacular drusen in both eyes ( Figure 2 ).
The third patient was first seen in our clinic at the age of 12 years for previously diagnosed Duane syndrome and a history of nasolacrimal duct obstruction. He was born at 35 weeks gestation, weighed 6 pounds, and suffered from neonatal sepsis. As an infant, the patient had upper gastrointestinal bleeding caused by esophagitis, delayed motor and language development, and hypotonia. He was diagnosed with mild cognitive delay, with an IQ score of 80. He had recurrent otitis media and blockage of both nasolacrimal ducts, requiring probing and irrigation at the age of 2 years and probing and tubing at the age of 5 years. In addition, he had crocodile tears syndrome and excessive drooling, believed to be caused by bulbar dysfunction. On physical examination, he had a long face, a thin upper lip, and a flat philtrum. His sisters have similar facial features, and his father has a very short upper lip. His eyes are prominent and his eyelashes are long. Patches of his hair are hypopigmented; he has preauricular pits, a bifid uvula, 4 missing teeth, and a coccygeal abnormality. He has a winged scapula, a large first toe on both feet, widely spaced nipples, and mild cubitus valgus deformity of the elbows. The patient has no hearing deficits, and his growth has been normal.
Because of the above abnormalities, genotyping was conducted by Signature Genomic Laboratories, LLC, using the Signature Chip microarray. This revealed a large chromosome 22 duplication extending from 22q11.1, the cat-eye locus, to 22q11.22, the DiGeorge syndrome 1 locus. This duplication was present in 57% of cells examined by FISH, and neither parent had the duplication, indicating a de novo mosaic pattern.
On presentation to our clinic, the patient reported good vision and occasional excessive tearing. His visual acuity was 20/15 in the right eye and 20/20 in the left eye with mild astigmatism in the left eye. He was unable to abduct either eye, and the globes retracted with narrowing of the palpebral fissures on adduction. There was no significant upshooting of either eye on adduction. The patient’s eyes were well aligned in primary gaze in both distance and near viewing, with no abnormal head posturing. Stereoacuity was normal, as was the remainder of the eye examination.
Discussion
Pathogenesis of Duane Syndrome
Duane Syndrome has Been Demonstrated to Result from an Sbsence of the Abducens Nerve and Nucleus in Some Cases, With Variable Innervation of the Lateral Rectus by the Oculomotor Nerve. The result is most commonly an absence of abduction with variable degrees of impairment of adduction. The simultaneous contraction of both medial and lateral rectus muscles on attempted medial gaze results in recession of the globe and narrowing of the palpebral fissures. On attempted lateral gaze, there is no muscle contraction and the palpebral fissures widen. There may be associated upshoot or downshoot of the affected eye with attempted adduction or abduction, and the patient may assume a head tilt to maintain single binocular vision.
There are 3 types of Duane syndrome that differ in patterns of dysinnervation and resulting clinical manifestations. Type I is the most common and is characterized by esotropia and complete limitation of abduction with little or no limitation of adduction. Type II is the least common and is characterized by exotropia in primary gaze, and complete limitation of adduction with little or no limitation of abduction. Type III involves marked or complete limitation of both adduction and abduction.
In most cases, Duane syndrome is sporadic. However, in approximately 10% of isolated cases, a family history of the condition is reported. In these cases, inheritance usually follows an autosomal-dominant pattern, and bilateral involvement is more likely than in sporadic cases. However, Duane syndrome has incomplete penetrance and variable expressivity, so some family members may not be affected, and familial cases may be unilateral and of varying severity. Multiple genes may be involved in the expression of the trait, and there may be interactions with environmental factors. Thirty percent to 50% of patients with Duane syndrome have other associated anomalies, usually in the ocular, auricular, skeletal, or neural structures. A few of these multisystem syndromes are discussed below. The significance of the association of Duane syndrome with other anomalies may be described by at least 3 distinct mechanisms. First, it has been hypothesized that a teratogenic insult at a certain point during development might affect multiple systems that are developing at the same time. In other cases, genetic mutations, amplifications, or deletions may cause Duane syndrome. Mutation of a gene important for the development of multiple systems, such as a transcription factor, may affect any or all of these systems. Alternatively, if genetic changes affect larger areas of chromosomes, additional systems may be directly affected by the chromosomal anomaly, so-called contiguous gene syndromes. In this report, we will focus on genetic causes of Duane syndrome and discuss the possible mechanisms involved in our 3 cases with chromosomal rearrangements/duplications.
Genetics of Duane Syndrome
Several Individuals Have Been Identified who Have Duane Syndrome and Abnormalities of Chromosome 8. In 1 patient, a deletion of chromosome 8q12.2-q21.2 resulted in a syndrome consisting of Duane syndrome, branchio-oto-renal (BOR) syndrome, hydrocephalus, and trapezius muscle aplasia. Individuals with BOR syndrome may have branchial cleft cysts or fistulae, preauricular pits, preauricular tags, malformations of the outer or middle ear, variable hearing loss, and hypoplastic kidneys. A second patient had Duane syndrome, microcephaly, mental retardation, and dysmorphic features associated with an insertion of 8q13-q21.2 onto 6q25 with a deletion of 8q13. Duane syndrome associated with facial dysmorphism, bilateral myopic astigmatism, congenital pendular nystagmus, macular hypoplasia, and other systemic abnormalities was found in a patient with trisomy 8 mosaicism. Another patient had Duane syndrome and hypoplastic external genitalia along with a reciprocal translocation of t(6;8)(q26;q13). The results of the above studies have led to the designation of the Duane retraction syndrome 1 (DURS1) locus, which is defined as Duane syndrome mapping to chromosome 8q13. The gene has not yet been identified.
Multiple studies have placed another gene for Duane syndrome within a region of chromosome 2 at 2q31 (DURS2). In the first of these, linkage analysis was performed in a family with autosomal-dominant Duane syndrome, placing the gene within a 17.8-cM region of 2q31. A second family with autosomal-dominant Duane syndrome was studied, and linkage was narrowed to an 8.8-cM region containing the homeobox D gene cluster. Two additional families were later identified, both mapping to the same region of 2q31. Finally, a DURS2 gene was identified as CHN1 using 4 patient pedigrees. CHN1 encodes α2-chimaerin, a RacGAP molecule known to be involved in neuronal signaling pathways and the development of corticospinal axons. Several of the CHN1 mutations identified in these 4 families with Duane syndrome resulted in increased α2-chimaerin activity. Furthermore, overexpression of α2-chimaerin in a chick in ovo model system caused aberrant cranial motor neuron development.
Abnormalities of chromosome 22 have been associated with Duane syndrome, in some cases in combination with urogenital abnormalities or cat-eye syndrome. Cat-eye syndrome is characterized by some combination of the following: preauricular pits and skin tags, ocular coloboma, congenital heart defects, anorectal malformations, urogenital anomalies, downward-slanting palpebral fissures, microphthalmia, hypertelorism, low-set ears, flattened nasal bridge, micro/retrognathia, epicanthic folds, skeletal anomalies, short stature, mild or moderate mental retardation, and strabismus. A family was studied where the father had only preauricular skin tags, but the son and daughter had Duane syndrome, renal agenesis, sensorineural deafness, and preauricular skin tags and sinuses. The daughter also had an absent uterus. All 3 had trisomy of a portion of chromosome 22, including the short arm and part of the long arm (22pter-q11). Several additional children have been found to have varying features of the cat-eye syndrome in conjunction with Duane syndrome and extra copies of portions of chromosome 22. In most of these cases, it was documented that the additional copies of chromosome 22 did not include 22q11.2, which is the site of the deletion that causes DiGeorge syndrome. DiGeorge syndrome is characterized by cardiac defects; abnormal facial features including wide-set eyes, low-set ears, micrognathia, and flat philtrum; thymus aplasia; cleft palate; hypocalcemia; and cognitive impairment. Another patient who had only a 22q11 microdeletion believed to represent DiGeorge syndrome was found to have a congenital laryngeal web, club feet, facial dysmorphism, and type II Duane syndrome on the left side.
When Duane syndrome is inherited with radial ray anomalies, the resultant condition is called Okihiro syndrome or Duane-radial-ray syndrome. Inheritance is autosomal dominant. Radial malformations affect the thumb and forearm. Additional findings may include sensorineural hearing loss and malformations of the thumb, forearm, vertebrae, lower extremities, heart, and kidneys. The gene for Okihiro syndrome was mapped to chromosome 20q, and mutations were identified within SALL4 , the gene for a zinc finger transcription factor.
There have been families and individual patients in whom Duane syndrome is associated with other clinical and chromosomal findings. A de novo deletion of 4q27-31 was found in a patient with bilateral Duane’s syndrome, bilateral blepharoptosis, and mild learning difficulties. A single case of bilateral Duane syndrome in conjunction with blepharophimosis-ptosis-epicanthus inversus syndrome (BPES) and FOXL2 mutation has been described. BPES is associated with mutations in FOXL2 in up to 75% of cases, and 20% to 25% of patients with BPES have either esotropia, exotropia, or hypertropia. FOXL2 is a transcription factor located on chromosome 3q23. Finally, a family with autosomal-dominant inheritance for Duane syndrome was studied for linkage to loci within chromosomes 4, 8, and 22, and no such linkage was found.
Significance of 3 New Cases of Duane Syndrome Associated With Chromosomal Duplications
We report 3 New Cases of Duane Syndrome with Documented Chromosomal Abnormalities. The first of these cases is consistent with a chromosome 2q13 duplication, potentially extending proximally as far as 2q11.1. This region does not overlap with the 2q31 locus containing CHN1 , a gene recently identified as causing Duane syndrome. However, it is notable that Duane syndrome caused by CHN1 mutation is the result of gene overexpression, and a duplication in our patient would also be expected to result in overexpression of genes important in causing Duane syndrome. The chromosome 2q13 duplication may be as large as 13.5 Mb and contain up to 160 genes. Several potential candidate genes reside within this chromosomal region. ARID5A (2q11.2) is involved in development, tissue-specific gene expression, and regulation of cell growth. SEMA4C (2q11.2) encodes a semaphorin, which is a class of secretory proteins important in neuron development, and POU3F3 (2q12.1) and NPAS2 (2q13) are transcription factors involved in neurogenesis. Very few patients with 2q duplications have been described in the past. Most of these previously described patients have duplications extending more distally than in our patient, and those that include 2q13 are most notable for being associated with a cleft palate.
The second patient has trisomy of a 33.75-Mb region of chromosome 10q24.2 to 10q26.3 as well as trisomy of the 3′ end of SLC2A10 at 20q13.12. Only 1 patient has been previously reported with partial trisomy of 10q. This patient has blepharophimosis, ptosis, severe mental retardation, and several other dysmorphic features secondary to a chromosome 4;10 translocation resulting in monosomy of 4q34.3qter and duplication of 10q25.1qter. This 10q duplication overlaps with the duplication present in our patient from 10q25.1 to 10q26.3, and the clinical findings of blepharophimosis, micrognathia, developmental delay, and mental retardation overlap as well. Several of the greater than 300 genes identified within our patient’s duplication are known to be associated with clinical syndromes that appear to be relevant to our patient’s presentation or to Duane syndrome. The first of these, FBXW4 at 10q24, is associated with split-hand and split-foot malformation type 3 (SHFM3). In addition to limb abnormalities, some patients have mental retardation and ectodermal and craniofacial findings. Interestingly, a gene involved in split-hand/foot malformation type 5 (SHFM5) is located at 2q31, the region also containing DURS1. The second gene of interest within this patient’s duplication is EMX2 at 10q26.1. This gene is associated with schizencephaly, a rare disorder of neuronal migration in which patients may have mental retardation, seizures, hypotonia, spasticity, inability to walk or speak, and blindness. Mutations in PAX2 (10q24.3-q25.1) cause papillorenal syndrome. Mutations in PITX3 (10q25) cause anterior segment ocular dysgenesis. PLEKHA1 (10q25.3-q26.2) and ARMS2 (10q26.13) are involved in susceptibility to age-related macular degeneration. These last 4 duplications might help explain the optic nerve hypoplasia, hypoplastic kidney, iris transillumination, and retinal drusen present in our patient. Many other genes within this large duplication may be involved in nervous system development as transcription factors or cell-signaling proteins.
Mutations of SLC2A10 , encoding the glucose transporter GLUT10, have been shown to cause arterial tortuosity syndrome. This syndrome is associated with arterial malformations, ischemic stroke, and Ehlers-Danlos syndrome, none of which appear to be present in our patient. However, some facial features found to be associated with arterial tortuosity syndrome, including micrognathia, elongated face, down-slanting palpebral fissures, blepharophimosis, and a beaked nose, are consistent with findings in our patient. No duplications of SLC2A10 have been identified in the past, so the significance of the small duplication in our patient remains unclear.
The third case is unique in that it is associated with a chromosome 22 duplication that includes both the cat-eye and DiGeorge syndrome regions. This patient has clinical findings most consistent with cat-eye syndrome, although there is some overlap with DiGeorge syndrome. There are nearly 100 genes included in the duplicated portion of chromosome 22. Many of these have been well studied and are believed to be important in embryogenesis. Any of these transcription factors and signaling proteins could potentially be involved in the pathogenesis of Duane syndrome.
To our knowledge, the chromosomal duplications in the first 2 patients described above have not been previously associated with Duane syndrome. The chromosome 22 duplication identified in the third patient overlaps with duplications previously described as including the cat-eye region, but also extends to the DiGeorge region. The duplications present in these 3 patients suggest that there may be several genes important for cranial nerve development that have not yet been identified as relevant to this process. As more patients with Duane syndrome undergo genetic testing, the regions containing candidate genes may be further narrowed. In vitro studies similar to those conducted by Miyake and associates will be necessary to confirm the involvement of individual candidate genes within these duplicated regions. By continuing to uncover new genetic causes of Duane syndrome, we may hope to gain a better understanding of cranial nerve development and the factors that are essential for normal nerve growth and differentiation.
Publication of this article was supported by an unrestricted grant from Research to Prevent Blindness, New York, New York, to the Cole Eye Institute (E.I.T.). Dr Traboulsi is a consultant and on the speakers’ bureau for Genzyme Corporation. Involved in design and conduct of the study (E.I.T.); collection, management, analysis, and interpretation of the data (S.S., E.I.T.); and preparation (S.S.), review, and approval of the manuscript (E.I.T.). This study was approved by the Institutional Review Board at the Cleveland Clinic.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


