Purpose
To standardize a novel submerged hydro-separation technique for Descemet membrane endothelial keratoplasty (DMEK) graft preparation from donor corneal tissues.
Design
Experimental study, laboratory investigation.
Methods
setting : The Veneto Eye Bank Foundation, Venice, Italy. study population : Fifty-four random human donor corneal tissues unsuitable for transplantation. intervention : Donor corneas were laid in a sterile basin partially filled with tissue culture medium. A 25 gauge needle with 1 mL mounted syringe was filled with the tissue culture medium. The needle (with bevel up) was bent to 90 degrees and was inserted in the posterior cornea initiating beneath the trabecular meshwork. It was further advanced toward the midperiphery, ensuring that only the bevel was inserted, considering it as a threshold of insertion. The liquid was injected with a medium to high pressure into the posterior stroma or in the Descemet membrane–stroma interface to create the bubble. The tissues were preserved for 7 days in tissue culture medium at 31°C. Parametrical, physiological and histological analyses were carried out. main outcome measures : Larger-diameter tissue, no tissue wastage, reproducibility, and preshipment evaluation.
Results
Complete detachment was achieved in all the cases without any tissue wastage. Average diameter of the excised graft was 10.80 (±0.28) mm and endothelial cell loss post preservation was 11.48%. Expression of tight junction protein and regular morphology was observed post preservation. No signs of cell apoptosis were seen. Histological analysis showed elimination of residual stroma in most of the cases.
Conclusions
The submerged hydro-separation method reduces tissue wastage. It allows preshipment evaluation, thus allowing a validated tissue to be transported from the eye banks to the surgeon. Because of the liquid interface, the peeling of the DMEK graft becomes easy for transplantation.
Human cornea is a transparent tissue with epithelium and Bowman layer as the anterior part, followed by the stroma, Descemet membrane, and endothelium. The anterior stroma is highly compact as compared to the posterior. Lamellar keratoplasties involve selective replacement of the diseased layers, as compared to penetrating keratoplasty (PK), which replaces a full-thickness corneal graft. Endothelial keratoplasty (EK) replaces the damaged or diseased posterior cornea with a healthy donor tissue. Although various techniques have been used to separate the Descemet membrane and the stroma, there is still a requirement of a standardized method. Currently, several techniques for Descemet membrane endothelial keratoplasty (DMEK) lenticule preparation from a donor cornea have been reported. It has been observed that many of these methods depend on personnel skill, and therefore the reproducibility is always a reason of concern, especially in a surgical theater. Thus, we set out to understand if the liquid bubble has any effect on the separation of the stroma and Descemet membrane using a submerged hydro-separation technique. Further, the technique was standardized to generate higher reproducibility and lower tissue wastage. The tissues were validated using a range of parameters as readouts in order to ensure a preseparated/prevalidated tissue ready for transportation and transplantation.
Materials and Methods
Donor Characteristics
A written consent from the donor’s family was obtained for the use of the corneal tissues for research purposes. The corneas were unsuitable for corneal transplantation owing to poor endothelial cell density (the Veneto Eye Bank Foundation standard is 2200 cells/mm 2 ) with an average of 2024.07 (±229.79) cells/mm 2 . However, the endothelial cell density with the corneas that were used for this study was >1800 cells/mm 2 and therefore the results can be extrapolated to the tissues that are used for transplantation purposes. The research tissues were used to check the histology, expression of markers, cell apoptosis, and cellular borders, for which the tissues have to be sacrificed; therefore, clinical-grade tissues were not used for this study. The average donor’s age was 64.96 (±5.58) years and the male-to-female ratio was 39:15. The corneas were preserved in tissue culture medium for approximately 2 weeks before the experiments.
Submerged Hydro-separation Method
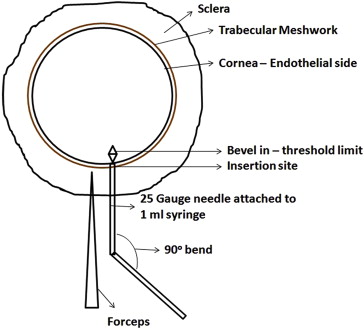
Fifty-four corneal tissues were submerged in a sterile petri plate half-filled with the tissue culture medium to keep the endothelium moist throughout the procedure. The tissue was held at the sclera with a sterile stainless steel forceps. A 25 gauge needle (90 degree angle bent with bevel up) connected to a 1 mL syringe filled with the tissue culture medium was inserted beneath the trabecular meshwork, as shown in Figure 1 . The needle was further moved radially beneath the endothelium in the posterior stroma or stroma–Descemet membrane interface until the bevel of the needle was completely inside the cornea, which was considered as the insertion threshold ( Figure 1 ). The schematic representation of the insertion site and the threshold limit is illustrated in Figure 2 . Tissue culture medium was injected into the tissue with medium to high pressure, enough to separate the Descemet membrane and the stroma. When injected into the posterior stroma, the liquid primarily filled the posterior stroma and later cleaved the stroma at its weak point, further entering into the stroma–Descemet membrane phase. A small, clear, visible bubble with the initiation of the process ensured that the procedure was accurate. The bubble was enlarged with an increased pressure to achieve a >10 mm diameter lenticule (measured using vernier calipers). The bubbles were prepared randomly by 2 operators to check the variability. The method is shown in Video clip 1 (Supplemental Material, available at AJO.com ).
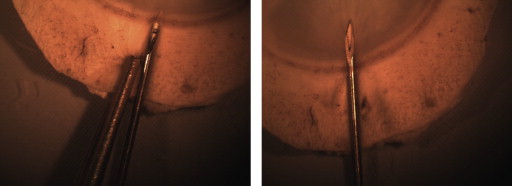
Preservation
The bubbles were partially collapsed ( Video clip 2 ; Supplemental Material, available at AJO.com ) before preservation to reduce any mechanical stretch/stress and were further preserved in the tissue culture medium for 7 days at 31°C in a humidity chamber.
Parametric Standards
The endothelium of all 54 samples was stained using trypan blue for 1 minute. The cornea was later washed in phosphate-buffered saline (PBS) and placed in a petri plate containing sucrose solution with the epithelium facing the lid. Five squares (1 mm 2 ) of 10 × 10 mm reticule (grid) inserted in the eyepiece of an inverted microscope were used for manual cell counting and mortality at 100× magnification. Descemet membrane and the stroma were separated using the above technique. Pre-bubble endothelial cell density and mortality were checked with an inverted microscope (Primovert; Zeiss, Milan, Italy) by masked observers. Variables such as the number of injections required to create the bubble (different sites of injection), quantity of the injected liquid, final diameter of the acquired bubble, post-bubble and postpreservation endothelial cell density, and mortality were recorded.
Alizarin Red Staining
Alizarin red staining was only used when it was difficult to identify the endothelial cell morphology using trypan blue and sucrose. The morphology (ie, pleomorphism, polymegathism, and hexagonality) of the cells was observed in 5 samples using alizarin red. After preservation of the tissues, the endothelium was stained (immersed) with alizarin red for 3.5 minutes, washed with PBS, and viewed at 100× magnification using an inverted microscope. In those cases when the cellular borders were not stained, restaining was performed with a higher exposure time of up to 5 minutes.
Tissue Fixation and Preparation for Cell Apoptosis and Immunostaining
To determine cell apoptosis and perform immunofluorescence-based analyses, 6 tissues were collected from each batch of two to check the reproducibility and variability. The preserved tissues were fixed in 4% paraformaldehyde (PFA) at 4°C overnight. The central DMEK lenticule was excised using a 9 mm Barron punch (Altomed, Tyne and Wear, England).
Immunostaining With Zonula Occludens-1 Staining
Twelve tissues were pretreated as described above and permeabilized with 0.5% Triton X-500 in PBS for 30 minutes. After blocking with 2% goat serum, the tissues were incubated overnight at 31°C in humidified atmosphere with a primary antibody (Zonula Occludens-1 [ZO-1], 1:500 dilution). The samples were incubated with goat anti-mouse fluorescein isothiocyanate (FITC)-conjugated secondary antibody in 20% goat serum for 3 hours at room temperature. Mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei. After each step, the cells were washed 3 times with 10× PBS. Cells were examined with an LSM 510-meta laser scanning microscope (Zeiss, Milan, Italy). Examination was performed under an ultraviolet light or by excitation at wavelengths of either 488 nm or 594 nm, and subsequent detection of the fluorescence was obtained.
Cell Apoptosis Using Terminal Deoxynucleotidyl Transferase Deoxyuridine Triphosphate Nick-End Labeling Assay
Cell apoptosis was performed as described in the manufacturer’s protocol for TACS 2 terminal deoxynucleotidyl transferase (TdT) diaminobenzidine (DAB) in situ apoptosis detection kit (Cat# 4810-30-K; Trevigen, Maryland, USA). Twelve samples were viewed at 10×, 20×, 40×, and 100× magnifications of an inverted microscope and the images were analyzed using ZEN (Zeiss, Milan, Italy) software.
Histology
After preservation, 20 tissues were fixed in 10% formalin. Periodic acid–Schiff (PAS) staining was performed on all the analyzed samples and viewed at 40× and 400× magnifications in order to check the variability and reproducibility in tissue selection and bubble performance along with stromal interference.
Scanning Electron Microscopy
The trephined lenticules with Descemet membrane side facing the top and histologic sections were immersed in 4% PFA at 4°C and left overnight. Five samples were then dehydrated with increasing concentrations of ethanol, exposing the samples for 30 minutes each (10%, 30%, 50%, 70%, 80%, and 95%). The samples were then placed in absolute alcohol and sent to the laboratory for scanning electron microscopy (SEM). Multiple cycles of liquid CO 2 at 0°C to 5°C followed by 32°C in a critical point dryer (CPD030; BAL-TEC AG, Balzers, Liechtenstein) were carried out. The samples were later attached to aluminum SEM stubs with graphite adhesives and sputter coated with gold (S150A-sputter coater Edwards, Crawley, England). The specimens were viewed on a JSM 6490 SEM (Jeol Ltd, Tokyo, Japan) to observe the Descemet membrane and the histologic sections of the lenticules at different magnifications to identify the smoothness and presence of any stromal residues.
Results
Pre-bubble endothelial cell density was 2024.07 (±229.79) cells/mm 2 with 1.67% (±6.82%) of trypan blue–positive cells. Double injections were required for 4 cases and triple for 2 cases, while the rest of the 48 corneas just needed a single injection to obtain the bubble. The average quantity of injected liquid was 0.57 (±0.29) mL (min, 0.3; max, 1.8). It was observed that the bubbles created with 0.3–0.5 mL showed less endothelial damage (9.52%) as compared to those with >0.8 mL (11.68%). The bubble with the right plane of cleavage could be achieved with 0.3–0.5 mL of liquid, and therefore no over-filling was carried out as it results in bubble burst. The average bubble diameter was found to be 10.80 (±0.28) mm (min, 10; max, 11). Post-bubble endothelial cell density was 2018.52 (±221.55) cells/mm 2 and the amount of trypan blue–positive cells was found to be 13.80% (±14.28%), as shown in Figure 3 . A higher percentage of trypan blue–positive cells were observed near the folds as compared to a very minimum that were scattered throughout. Postpreservation endothelial cell density was found to be 1791.66 (±238.92) cells/mm 2 with 0.80% (±1.61%) trypan blue–positive cells, as shown in Figure 4 . The mortality was reduced as the neighboring cells expand and cover the space of the dead cells. Therefore, the average endothelial cell loss recorded was 11.48% starting from the fresh cornea to postpreservation.
Alizarin red staining showed a regular morphologic display of the cells post preservation, thus suggesting that, although some cells lost their viability, the morphology was reorganized after preservation of the tissues in tissue culture medium after 7 days ( Figure 5 ) with minor polymorphism. Apart from morphology, the intercellular tight junctions showed expression of ZO-1 protein, which leads to the conclusion that the tight junctions were preserved even after mechanical stress. DAPI fluorescent nuclei staining further confirmed the presence of the cells on the DMEK tissue ( Figure 6 ). Even though a stress is conferred on the cells during bubble formation, preserved tissues showed expression of these proteins. Tissue culture medium has been reported to be toxic after preservation of the tissues for over 3 weeks. However, our study did not show any cellular apoptosis on the DMEK tissues prepared using liquid bubble and preserved in tissue culture medium for a week. To confirm these data, positive controls were induced with apoptosis and the difference between the control and samples was recorded, as shown in Figure 7 .



