Purpose
To screen primary congenital glaucoma patients in the United States for sequence variants within the CYP1B1 , LTBP2 , and MYOC genes using Sanger and whole exome sequencing.
Design
Retrospective case-control study.
Methods
Fifty-seven primary congenital glaucoma patients (47 families), 71 unaffected family members of the primary congenital glaucoma probands, and 101 healthy unrelated individuals were recruited from a single institution. Sanger sequencing of the primary congenital glaucoma gene, CYP1B1 , was performed on 47 proband deoxyribonucleic acid samples. Simultaneously, whole exome sequencing was conducted on 3 families, each including more than 1 affected individual. Concurrently, 33 of 47 primary congenital glaucoma probands with extended family deoxyribonucleic acid samples were screened for LTBP2 and MYOC gene mutations. Exome-sequenced variations were validated by additional Sanger sequencing to confirm segregation of filtered disease-causing single nucleotide variations.
Results
Seven primary congenital glaucoma families (14.9%) manifested disease phenotypes attributable to CYP1B1 mutations. One primary congenital glaucoma family possessed homozygous mutant alleles, whereas 6 families carried compound heterozygous mutations. Five novel combinations of compound heterozygous mutations were identified, of which 2 combinations were found with whole exome sequencing. No disease-causing mutations within the LTBP2 and MYOC genes were discovered.
Conclusions
This study analyzed CYP1B1 , LTBP2 , and MYOC mutations in a cohort of primary congenital glaucoma patients from the United States, applying whole exome sequencing as a complementary tool to Sanger sequencing. Whole exome sequencing, coupled with Sanger sequencing, may identify novel genes in primary congenital glaucoma patients who have no mutations in known primary congenital glaucoma genes.
Primary congenital glaucoma (OMIM 231300) is a rare but devastating eye disease. It is characterized by congenital elevation of intraocular pressure (IOP) resulting from significantly reduced aqueous outflow through a malfunctioning trabecular meshwork. The raised IOP results in progressive optic nerve damage, potentially leading to blindness. Other clinical features include buphthalmos, corneal edema and opacification with rupture of the Descemet membrane, thinning of the anterior sclera, iris atrophy, and an anomalously deep anterior chamber. The disease presents classically in infants and toddlers with the symptoms of epiphora, blepharospasm, and photophobia. Typically, the diagnosis is made within the first year of life.
The incidence of primary congenital glaucoma varies based on ethnicity and ranges from as high as 1 in 1250 persons in the Gypsy population of Slovakia to as low as 1 in 18,500 to 1 in 30,000 persons in Western populations. The disease accounts for 5% of childhood blindness and approximately 18% of children in blind institutions worldwide. It can occur in both sporadic and familial patterns. Inheritance usually is autosomal recessive in familial cases, with increased association with consanguinity. Three genetic loci—GLC3A (OMIM 231300), GLC3B (OMIM 600975), and GLC3C (OMIM 613085)—have been identified by linkage analyses in large multigenerational pedigrees. A new locus, GLC3D (OMIM 613086), on 14q24, recently was characterized. To date, 3 genes have been implicated, and include cytochrome P450, subfamily I, polypeptide 1 ( CYP1B1 ; NM_000104.3 ), latent transforming growth factor β binding protein 2 ( LTBP2 ; NM_000428.2 ), and myocilin ( MYOC ; NM_000261.1 ).
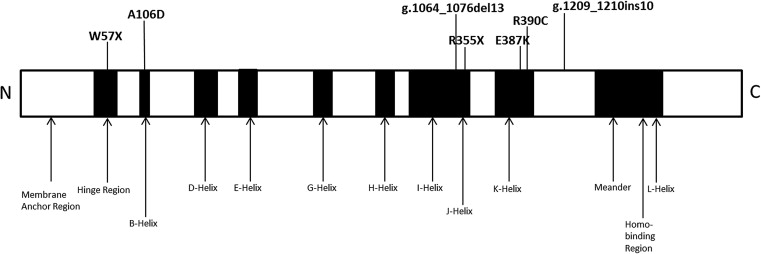
The CYP1B1 gene was the first gene in which mutations were found to cause primary congenital glaucoma. It is located on chromosome 2p22–p21 within the GLC3A locus. Its protein oxidizes compounds important to eye structure and function, including steroids, retinoids, arachidonate, and melatonin. Studies have demonstrated its expression in fetal and adult ciliary body and neuroepithelium, but not in the trabecular meshwork. It is speculated that the enzyme metabolizes an unknown molecule critical to eye development. The proportion of patients with pathogenic CYP1B1 sequence variants varies with ethnicity, ranging from 100% in Slovakian Romas to 20% in Japanese individuals.
LTBP2 is located on chromosome 14q24 within the GLC3D locus. In nonocular tissues, LTBP2 is involved in tissue repair and cell adhesion. Ocular expression studies have determined its presence in both the trabecular meshwork and ciliary processes. Several studies have shown that LTBP2 mutations are associated with secondary glaucoma. The role of LTBP2 in primary congenital glaucoma remains unclear, because some studies did not discover any mutations within the gene, but null mutations have been found in consanguineous Slovakian Roma, Pakistani, and Iranian families.
MYOC also is associated with juvenile and primary open-angle glaucoma and is located on chromosome 1q24.3–q25.2. Pathogenic MYOC variant proteins can alter the trabecular meshwork and ciliary body architecture, obstructing the outflow and increasing IOP. Disease-causing MYOC sequence variants, in the presence or absence of CYP1B1 mutations, have been reported in families with primary congenital glaucoma.
In this study, we sought to determine CYP1B1 , LTBP2 , and MYOC sequence variations in a large multiethnic cohort of families and cases with primary congenital glaucoma in the United States. In most instances, Sanger sequencing was performed initially. In select families, whole exome sequencing was used as the initial screening tool for genetic mutations. Compared with other sequencing methods, whole exome sequencing technology has matured in recent years to emerge as an accurate and efficient means for detecting both known and novel genetic mutations in a range of ocular diseases, including corneal dystrophy and inherited retinal disorders. To our knowledge, this is the first study to perform whole exome sequencing analysis in a primary congenital glaucoma cohort. Sanger sequencing was performed to confirm identified mutations.
Methods
Subjects
The participants were recruited at the Duke University Eye Center, from the clinical practices of 2 of the authors (S.F.F. [a majority] and T.L.Y.). The retrospective case-control study was approved by the Duke University Institutional Review Board and adhered to the tenets of the Declaration of Helsinki. The Ophthalmic Genetics Institutional Review Board (protocol no. 00008040) approved the recruitment of individuals and family members with hereditary developmental ophthalmologic disorders, the collection of blood or cheek cell samples for deoxyribonucleic acid (DNA) extraction, the screening for genetic mutations, and the making of genotype and phenotype correlations. Written informed consent for study participation was obtained from the subject or the subject’s parents, as appropriate. The study also complied with the Health Insurance Portability and Accountability Act. Primary congenital glaucoma was defined by the following characteristics: (1) age of onset of 3 years of younger; (2) increased corneal diameter of more than 10 mm accompanied by either corneal edema, Haab striae, or both; and (3) increased IOP of more than 21 mm Hg, optic nerve cupping of more than 0.4, asymmetry of more than 0.2, or a combination thereof. Any patient with other ocular abnormalities or systemic conditions, other than iris stromal hypoplasia, was excluded from the study. Excluded ocular anomalies included posterior embryotoxon, corectopia, cataract, Axenfeld Reiger syndrome, Peters anomaly, and aniridia. A questionnaire regarding family and medical history was completed by the subject or the subject’s parents, or both. Blood or saliva samples were collected from the subject and family members; genomic DNA was extracted using AutoPure LS DNA Extractor and PUREGEN reagents (Gentra Systems Inc, Minneapolis, Minnesota, USA).
A total of 47 unrelated primary congenital glaucoma probands, 10 additional affected siblings, and 71 unaffected family members of the probands were enrolled in this study. A second dataset of 101 healthy unrelated individuals with normal ophthalmic examinations and no family history of hereditary ocular diseases were recruited randomly as control subjects. The ethnic distribution of this control group was matched to our primary congenital glaucoma cohort.
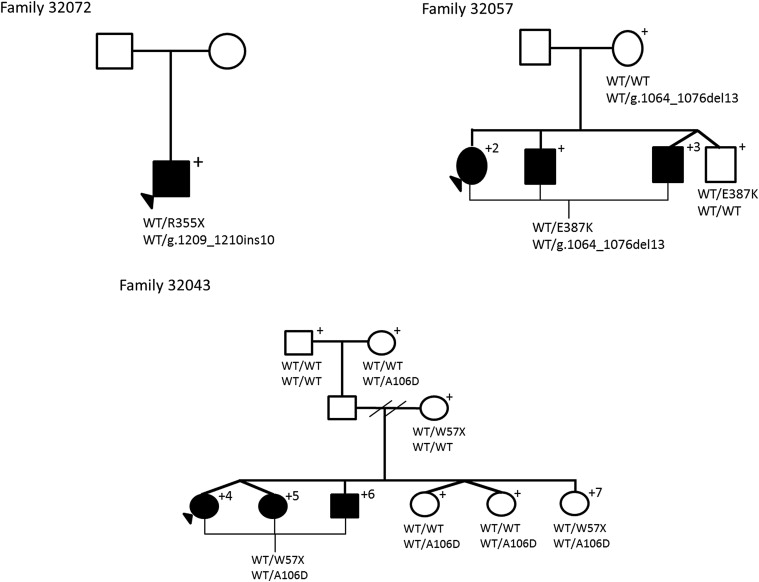
Before DNA sample submission of 3 primary congenital glaucoma families for whole exome sequencing, Sanger sequencing was conducted on family 32043 to search for candidate gene mutations. However, only known combinations of compound heterozygous mutations were assessed. This family demonstrated a novel CYP1B1 compound heterozygous mutation revealed by whole exome sequencing, and Sanger sequencing was repeated to confirm the finding. In total, DNA samples of 7 individuals from 3 families underwent whole exome sequencing.
Whole Exome Sequencing
Because of resource availability and the timing of experiments, 2 sequencing service providers were used: the Hudson Alpha Institute, Huntsville, Alabama, and the Beijing Genomics Institute, Hong Kong, China, with different library captures and coverage depths for exome sequencing.
SeqCap EZ Human Exome Library version 2.0 exome sequencing
DNA samples (7 μg) of 2 affected individuals from family 32057 and of 1 affected individual from family 32071 were submitted to the Hudson Alpha Institute for exome sequencing ( Figures 1 and 2 ). The DNA was sheared randomly between 150 to 200 base pairs (bp), and adapters were ligated to both ends of the fractions. Biotinylated ribonucleic acid capture probes from the SeqCap EZ Human Exome Library version 2.0 (Roche NimbleGen, Madison, Wisconsin, USA) were hybridized to the prepared DNA fractions. The hybrid-selected enriched output library was amplified by polymerase chain reaction before targeting the sequencing depth of ×50. The SeqCap EZ Human Exome Library version 2.0 was used as target covering of approximately 36.5 Mb of the human genomic regions of consensus coding sequence exons.
Agilent SureSelect 38-Mb exome sequencing
Seven-μg DNA samples of 3 affected siblings and 1 unaffected sibling from family 32043 were submitted to the Beijing Genomics Institute for independent processing ( Figure 3 ). Exome libraries were generated using the Agilent SureSelect 38-Mb kit (Agilent Technologies, Santa Clara, California, USA), which captured 1.22% of human genomic regions covering consensus coding sequence exons. Samples were processed similarly as above and were sequenced to obtain an average exome coverage depth of ×30.
Bioinformatics: Read Mapping and Quality Filtering
The paired reads were aligned first to the National Center for Biotechnology Information GRCh37 lite reference genome ( ftp://ftp.ncbi.nih.gov/genbank/genomes/Eukaryotes/vertebrates_mammals/Homo_sapiens/GRCh37/special_requests ), with the Burrows-Wheeler Alignment tool and Sequence Alignment/Map tools ( http://samtools.sourceforge.net/ ). All aligned reads in the merged binary alignment and map file were processed with the Genome Analyzer Toolkit (version 4333; Broad Institute, Cambridge, Massachusetts, USA). Consensus calling was performed with the Genome Analyzer Toolkit framework to obtain single nucleotide variants (SNVs). SNVs were filtered by quality and depth, minimum total read depth (coverage), and occurrence of other SNVs or microinsertions or microdeletions (microindels) close to the variant position. SNVs passing these filter thresholds were annotated with information from the University of California, Santa Cruz genome annotation database ( http://genome.ucsc.edu/index.html ), 1000 Genomes Project ( http://www.1000genomes.org/ ), consensus coding sequence ( http://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi ), Ensembl ( http://www.ensembl.org ), RefSeq ( http://www.ncbi.nlm.nih.gov/RefSeq/ ), MirBase ( http://www.mirbase.org/ ), and EntrezGene ( www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene ).
To remove common SNVs, variants present in dbSNP132 or the 1000 Genomes database were removed, unless the minor allele frequency was less than 3% or unknown. Subsequently, only coding nonsynonymous variants or splice site mutations were analyzed. Two programs, SIFT ( http://sift.jcvi.org/ ) and PolyPhen2 (version 2.1.0 r367; http://genetics.bwh.harvard.edu/pph2/ ), were used to predict the functional impact of the sequence variants on the gene-encoded protein.
Variant Screening Using Next-Generation Sequencing
Variant Call Format files generated were used for analysis, reviewing all variants within CYP1B1 , MYOC , and LTBP2 genes. Concurrently, binary alignment and map files were integrated into the Integrative Genomics Viewer ( http://www.broadinstitute.org/igv/ ) to confirm coverage by visualization. Previously reported pathogenic mutations or novel variants identified were Sanger sequenced for confirmation.
Polymerase Chain Reaction and Deoxyribonucleic Acid Sequencing
Concurrent with or after whole exome sequencing, Sanger sequencing was carried out on all affected individuals to screen for CYP1B1 sequence variants. Sequencing of the LTBP2 and MYOC genes was performed selectively on 33 affected individuals for whom DNA samples of additional family members were available to confirm cosegregation with disease status.
The primers were designed using the Primer3 program ( http://frodo.wi.mit.edu/ ; Supplemental Table 1 , available at AJO.com ). Standard polymerase chain reaction conditions were run to amplify all exons, intron and exon boundaries, and untranslated regions. Amplicons were visualized after electrophoresis on a 2% agarose gel and were purified with Quickstep 2 SOPE Resin (Edge BioSystems, Gaithersburg, Maryland, USA). Using ABI BigDye chemistry (Applied Biosystems Inc, Foster City, California, USA), Sanger sequencing was carried out on the polymerase chain reaction amplicons and was processed through an automated ABI 3730 Sequencer (Applied Biosystems, Inc). The sequences were scrutinized for variations alongside reference sequences from the University of California, Santa Cruz genome browser website ( http://genome.ucsc.edu/index.html ) using Sequencher 5.0 software (Gene Codes, Ann Arbor, Michigan, USA). Sanger sequencing subsequently was performed on unaffected family members of cases with identified sequence variants and on 101 unaffected control samples.
| Primary Congenital Glaucoma Family | Individual Number | Mutation | dbSNP Identification Data | Base Pair Location a | Next-Generation Sequencing Coverage b |
|---|---|---|---|---|---|
| 32071 | 1 | p.Trp57* | rs72549387 | 38302361 | ×4 |
| 10-bp insertion | rs72466463 | 38298287_38298288insGGTGGCATGA | ×19 | ||
| 32057 | 2 | p.Glu387Lys | rs55989760 | 38298338 | ×11 |
| 13-bp deletion | rs72549380 | 38298421_38298433delTCTGCCTGCACTC | ×6 | ||
| 3 | p.Glu387Lys | rs55989760 | 38298338 | ×16 | |
| 13-bp deletion | rs72549380 | 38298421_38298433delTCTGCCTGCACTC | ×7 | ||
| 32043 | 4 | p.Trp57* | rs72549387 | 38302361 | ×13 |
| p.Ala106Asp | Novel | 38302215 | ×11 | ||
| 5 | p.Trp57* | rs72549387 | 38302361 | ×7 | |
| p.Ala106Asp | Novel | 38302215 | ×8 | ||
| 6 | p.Trp57* | rs72549387 | 38302361 | ×9 | |
| p.Ala106Asp | Novel | 38302215 | ×15 | ||
| 7 (Unaffected) | p.Trp57* | rs72549387 | 38302361 | N/A | |
| p.Ala106Asp | Novel | 38302215 | N/A |
b Number of reads present at the particular location using Variant Call Format files or visualization using Integral Genomics Viewer.
Results
A total of 47 families with 1 or more individual(s) affected by primary congenital glaucoma were studied: 26 (55.3%) subjects were male, whereas 21 (44.7%) subjects were female. With respect to ethnicity, 27 (57.5%) were white, 16 (34.0%) were black, 2 (4.3%) were Hispanic, 1 (2.1%) was Asian, and 1 (2.1%) had a multiracial background. Forty (85.1%) of the 47 families had only 1 affected individual.
DNA samples of 3 affected individuals from 2 families were exome sequenced (2 from family 32057, 1 from family 32071) using the SeqCap EZ Human Exome Library version 2.0 at ×50 coverage. An average of 85 million reads overlapped with targeted positions, with a mean coverage of ×77. On average, 96% of the targeted exons had at least ×1 coverage, and an average of 88% of the exons were captured at ×10 coverage depth ( Supplemental Table 2 , available at AJO.com ). Gene-specific coverage of CYP1B1 , LTBP2 , and MYOC demonstrated mean coverage depths of ×50, ×33, and ×51, respectively. In family 32071, a nonsense mutation (p.Trp57*) and a 10-bp insertion (rs72466463) were identified as a compound heterozygous state in the CYP1B1 gene ( Figure 1 and Table 1 ). In family 32057, a missense mutation (p.Glu387Lys) and a 13-bp deletion (rs72549380) in a compound heterozygous state were identified in both affected individuals in the CYP1B1 gene ( Figure 2 and Table 1 ).
| Exon | rs Number | Function | Protein Residue (Amino Acid) Change |
|---|---|---|---|
| 2 | rs10012C→G | CNS | p.R48G |
| 2 a | rs72549387G → A | CNS | p.W57X |
| 2 a | (no rs number) C→A | CNS | p.A106D |
| 2 | rs1056827G→T | CNS | p.A119S |
| 2 | rs9341247C→A | CS | p.G188G |
| 2 | rs9341249G→C | CS | p.V243V |
| 3 a | rs72549381C→T | CNS | p.R355X |
| 3 a | rs55989760G→A | CNS | p.E387K |
| 3 a | rs148542782C→T | CNS | p.R390C |
| 3 | rs1056836C→G | CNS | p.L432V |
| 3 a | rs72466463 | 10-bp insertion | Frameshift |
| 3 a | rs72549380 | 13-bp deletion | Frameshift |
| 3 | rs4986888C→G | CNS | p.A443G |
| 3 | rs1056837T→C | CS | p.D449D |
| 3 | rs1800440A→G | CNS | p.N453S |
a Sequence variants determined to be associated with primary congenital glaucoma in this study.
DNA samples of 3 affected individuals and 1 unaffected individual from family 32043 were exome sequenced using the Agilent SureSelect 38-Mb library capture system with a target depth of ×30 coverage ( Supplemental Table 2 , available at AJO.com ). On average, more than 95% of the targeted exons were covered with at least ×1 coverage, and an average of 74% of the exons were captured at ×10 in DNA samples of 4 individuals ( Supplemental Table 2 , available at AJO.com ). The average gene-specific coverage depths for CYP1B1 , LTBP2 , and MYOC were ×16, ×27, and ×45, respectively. Within family 32043, Variant Call Format alone identified a nonsense mutation p.Trp57* (rs72549387) and a missense mutation p.Ala106Glu in heterozygous states in affected individuals 4, 5, and 6 ( Figure 2 and Table 1 ). Both mutations were located within the CYP1B1 gene. Visualization using the Integrative Genomics Viewer determined unaffected individual 7 to have too low of a coverage depth to identify variants at the same positions.
Summary of CYP1B1 Sequence Variations
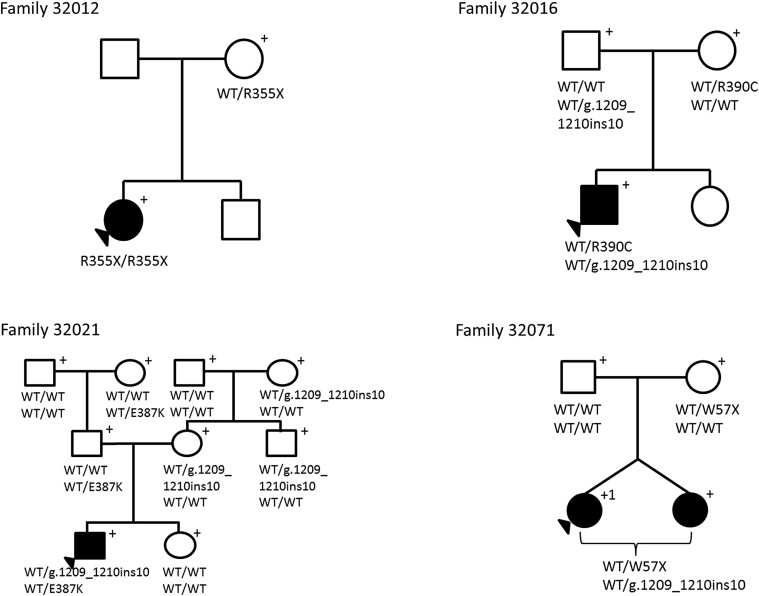
Fifteen coding sequence variants were identified within the CYP1B1 gene ( Table 2 ). Six variants were located in exon 2, whereas 9 variants were in exon 3. Eleven variants were nonsynonymous, whereas 2 were synonymous, comprising 6 transitions and 7 transversions. The transitions were 2 nonsense variants: c.171G→A (p.Trp57*, rs72549387), c.1063C→T (p.Arg355*, rs72549381), and 4 missense variants: c.1159G→A (p.Glu387Lys, rs55989760), c.1168C→T (p.Arg390Cys, rs148542782), c.1347T→C (p.Ala443Gly, rs1056837), and c.1358A→G (p.Asn453Ser, rs1800440). The 7 transversions were c.142C→G (p.Arg48Gly, rs10012), c.317C→A (p.Ala106Asp, no rs identification), c.355G→T (p.Ala119Ser, rs1056827), c.564C→A (p.Gly188Gly, rs9341247), c.729G→C (p.Va1243Val, rs9341249), c.1294C→G (p.Leu432Val, rs1056836), and c.1328C→G (p.Ala443Gly, rs4986888). There was a 10-bp insertion c.1209_1210insTCATGCCACC (rs72466463) and a 13-bp deletion c.1064_1076delGAGTGCAGGCAGA (rs72549380), and both resulted in sequence frameshifts. Seven of the 47 primary congenital glaucoma families carried known disease-causing CYP1B1 mutations determined to be absent in the 101 control subjects ( Figure 3 ). Of the 7 families, an affected black individual had a homozygous change, whereas affected individuals from 1 black and 5 white families had compound heterozygous changes. To our knowledge, 5 of the 6 combinations of compound heterozygous variants are novel ( Table 3 ). Clinically, members of all 7 families had severe disease phenotypes that required trabeculectomy or placement of aqueous drainage devices within 6 months of diagnosis because of poorly controlled IOP.
| Primary Congenital Glaucoma Family | Homozygous/Heterozygous | CYP1B1 Amino Acid Changes | Combination Previously Reported |
|---|---|---|---|
| 32012 | Homozygous | p.Arg355*/p.Arg355* | Yes |
| 32016 | Compound heterozygous | p.Arg390Cys/c.1209_1210insTCATGCCACC | No |
| 32021 | Compound heterozygous | p.Glu387Lys/c.1209_1210insTCATGCCACC | No |
| 32071 | Compound heterozygous | p.Trp57*/c.1209_1210insTCATGCCACC | Yes |
| 32072 | Compound heterozygous | p.Arg355*/c.1209_1210insTCATGCCACC | No |
| 32057 | Compound heterozygous | p.Glu387Lys/c.1064_1076delGAGTGCAGGCAGA | No |
| 32043 | Compound heterozygous | p.Trp57*/p.Ala106Asp | No |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


