Purpose
To investigate the presence of TBK1 copy number variations in a large, well-characterized Australian cohort of patients with glaucoma comprising both normal-tension glaucoma and high-tension glaucoma cases.
Design
A retrospective cohort study.
Methods
DNA samples from patients with normal-tension glaucoma and high-tension glaucoma and unaffected controls were screened for TBK1 copy number variations using real-time quantitative polymerase chain reaction. Samples with additional copies of the TBK1 gene were further tested using custom comparative genomic hybridization arrays.
Results
Four out of 334 normal-tension glaucoma cases (1.2%) were found to carry TBK1 copy number variations using quantitative polymerase chain reaction. One extra dose of the TBK1 gene (duplication) was detected in 3 normal-tension glaucoma patients, while 2 extra doses of the gene (triplication) were detected in a fourth normal-tension glaucoma patient. The results were further confirmed by custom comparative genomic hybridization arrays. Further, the TBK1 copy number variation segregated with normal-tension glaucoma in the family members of the probands, showing an autosomal dominant pattern of inheritance. No TBK1 copy number variations were detected in 1045 Australian patients with high-tension glaucoma or in 254 unaffected controls.
Conclusion
We report the presence of TBK1 copy number variations in our Australian normal-tension glaucoma cohort, including the first example of more than 1 extra copy of this gene in glaucoma patients (gene triplication). These results confirm TBK1 to be an important cause of normal-tension glaucoma, but do not suggest common involvement in high-tension glaucoma.
Glaucomas are a group of eye diseases with a common feature of progressive irreversible degeneration of the optic nerve with corresponding loss of the peripheral visual field. Glaucomas are the leading cause of irreversible blindness worldwide, and primary open-angle glaucoma is the most prevalent subtype worldwide. The main risk factor for glaucoma is elevated intraocular pressure; however, approximately 20%–50% of all primary open-angle glaucoma cases present with normal intraocular pressure range (10–21 mm Hg) and are termed normal-tension glaucoma.
The genetic contribution to primary open-angle glaucoma is well documented. Around half of all primary open-angle glaucoma patients have a positive family history, and first-degree relatives of primary open-angle glaucoma patients have an approximately 9-fold increased risk of developing glaucoma. The first gene identified to be associated with familial normal-tension glaucoma was Optineurin ( OPTN ) in the GLC1E region on chromosome 10p15-14. Subsequent studies reported that mutations in OPTN cause 1%–2% of primary open-angle glaucoma or normal-tension glaucoma. Despite several studies on the OPTN gene, its exact role in causing primary open-angle glaucoma remains elusive.
Recently, a novel genetic locus (GLC1P) on chromosome 12q14 was reported to be linked to normal-tension glaucoma in an African-American pedigree. A duplication that spans the TANK binding kinase 1 ( TBK1 ) gene was subsequently detected in this pedigree, as well as in 2 out of 153 unrelated normal-tension glaucoma subjects from Iowa (1.5%), 1 out of 252 unrelated Japanese normal-tension glaucoma patients (0.4%), and 1 out of 96 unrelated patients from New York (1.0%). These data suggest that abnormal TBK1 dosage (duplication) causes normal-tension glaucoma in these patients. The association between copy number variations of the TBK1 gene and normal-tension glaucoma is supported by several additional observations. First, copy number variations are known to be involved in influencing gene expression and are risk factors for primary open-angle glaucoma ( GALC gene) and Axenfeld-Rieger syndrome ( FOXC1 gene), as well as a number of diseases such as HIV dementia complex, autism, and Alzheimer disease. Second, TBK1 is specifically expressed in the ganglion cells and the nerve fiber layer of the human retina, which are involved in the pathogenesis of glaucoma. Third, OPTN binds the TBK1 protein, particularly in the presence of the recurrent severe glaucoma-causing mutation E50K in the OPTN gene. Interestingly, 3 known normal-tension glaucoma genes ( TBK1 , OPTN , and TLR4 ) each encode proteins that directly interact with each other in a biological pathway that activates autophagy, a process by which intracellular materials (eg, proteins, organelles, or pathogens) are degraded. Together these data further implicate the role of the TBK1 gene in the pathogenesis of normal-tension glaucoma.
In this study, we aimed to investigate the presence of copy number variations of the TBK1 gene in unrelated normal-tension glaucoma cases and unaffected controls recruited from the Australian population. We also explored the presence of the gene copy number variations in patients with high-tension glaucoma, thus attempting to define an overall contribution of TBK1 copy number variations to glaucoma blindness.
Methods
Approval of this retrospective cohort study was obtained from the Southern Adelaide Clinical Human Research Ethics Committee. This study has been conducted in accordance with the Declaration of Helsinki and its subsequent revisions. The committee prospectively approved the recruitment of individuals and family members with primary open-angle glaucoma and its subtypes, the collection of blood or saliva samples for deoxyribonucleic acid extraction, the screening for genetic mutations, the data analysis, and the making of genotype and phenotype correlations. Written informed consent was obtained from each individual to participate in this study. Recruitment was conducted through the Australian and New Zealand Registry of Advanced Glaucoma. The unaffected control cohort was collected from retirement villages in Adelaide, South Australia, as previously described.
Each participant was examined by his or her specialist and received a complete eye examination, including slit-lamp examination of the anterior chamber, gonioscopy, measurement of central corneal thickness (CCT), visual acuity, intraocular pressure, fundus examination with special attention to optic disc health and size, and automated perimetry. The diagnosis of glaucoma followed the definition of the International Society of Geographical and Epidemiological Ophthalmology described by Foster and associates, with optic nerve damage and corresponding visual loss detected in at least 1 eye. Patients recruited in the study and identified as having normal-tension glaucoma followed the same criteria described by Fingert and associates (intraocular pressure less than or equal to 21 mm Hg in both eyes, unadjusted for CCT). High-tension glaucoma patients were diagnosed with intraocular pressure greater than 21 mm Hg in at least 1 eye, along with glaucomatous optic nerve and visual field damage. Patients diagnosed with advanced glaucoma presented with either fixation involving visual field loss (at least 2 of the 4 central fixation squares having a pattern standard deviation of less than 0.5% on a reliable Humphrey 24-2 field) or severe global field loss at baseline (mean deviation of less than −22 dB) in at least 1 eye. Family members of TBK1 copy number variation carriers were recruited when available. The controls had no evidence of glaucomatous optic nerve damage, intraocular pressure of less than or equal to 21 mm Hg, and no family history of glaucoma, and were slightly older than cases by design for this aging disease. The study was first conducted using a total of 334 unrelated patients with normal-tension glaucoma and 254 unaffected controls. Sixty-three percent of patients (n = 212) had advanced normal-tension glaucoma, while the remainder (n = 122) had less severe (nonadvanced) normal-tension glaucoma. A positive family history of glaucoma was present in 133 patients (40%).
Venous blood samples were obtained from the participants for the study. Genomic DNA was extracted from peripheral whole blood using the QiaAmp Blood Maxi Kit (Qiagen, Valencia, California, USA). DNA from each subject was tested for TBK1 duplications using TaqMan Copy Number Assays (Life Technologies, Carlsbad, California, USA). The segment of the TBK1 gene was amplified in 4 replicates for each DNA sample. The experiment was conducted using the StepOne Plus real-time polymerase chain reaction instrument, which quantitates the gene of interest, normalized to an endogenous reference gene (RNase P) known to be present in 2 copies in a diploid genome. Evaluation of the copy number of genomic DNA targets was performed using the CopyCaller 2.0 software (Life Technologies) with default settings. For detailed mapping of duplication events, patients with detected TBK1 duplications were analyzed using custom 8x60K SurePrint G3 Human custom comparative genomic hybridization microarrays (Agilent, Santa Clara, California, USA) that interrogated over 55 000 probes in the GLC1P locus that spans 9.5 Mbp between rs12227270 and rs7488555 on chromosome 12q14, using the manufacturer’s protocol.
To further explore the relationship between 2 apparently unrelated individuals with an identical duplication, we analyzed the haplotypes surrounding the duplication region. The 3 carriers with primary open-angle glaucoma were also part of a previously reported genome-wide association scan (GWAS). Along with 590 other participants with primary open-angle glaucoma, they were genotyped on the Omni1 array (Illumina, San Diego, California, USA). The most likely haplotype pair across the duplication region (chr12:64173733–65613733, hg19) in each participant in the GWAS was estimated using Beagle 3.3.2 ( http://faculty.washington.edu/browning/beagle/beagle.html ). The haplotypes across the whole region were visually compared between patients AG624 and AG724. The haplotypes for AG604 with a different duplication were also compared.
Mutation screening of TBK1 was performed on 95 unrelated cases with high-tension glaucoma, 100 unrelated cases with normal-tension glaucoma, and 104 unaffected unrelated controls from Australia. Exome capture was performed using the SureSelect system (Agilent) and paired-end libraries were sequenced on an Illumina HiSeq 2000 by Macrogen Inc (Seoul, South Korea). Reads were mapped to the human reference genome (hg19) using BWA ( http://bio-bwa.sourceforge.net/ ), and duplicates were marked and removed using picard. Variants were called using SAMtools and annotated with ANNOVAR ( http://www.openbioinformatics.org/annovar/ ). Variants were described according to the recommendations of the Human Genome Variation Society ( http://www.hgvs.org/ ) and referenced against the NHLBI Exome Variant Server ( http://evs.gs.washington.edu/EVS/ [July 2014]), 1000 Genomes, and dbSNP v138 databases ( http://www.ncbi.nlm.nih.gov/snp ).
Results
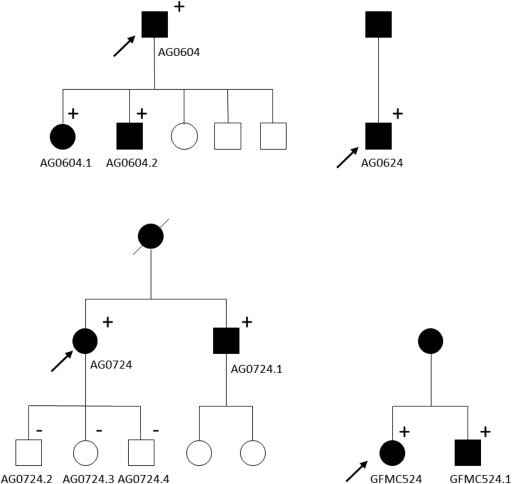
TBK1 copy number variations were detected in 4 of 334 Australian cases with normal-tension glaucoma (1.2%) using quantitative polymerase chain reaction assays ( Figure 1 ). Three unrelated probands, GFMC524, AG604, and AG624, were found to have 3 copies of the gene (1 extra dose), while AG724 participant was found to carry 4 total copies of TBK1 (2 extra doses). No copy number variations were detected in any of the unaffected controls. This rate is similar to previously published data where overlapping copy number variations were found in 1.3% of white normal-tension glaucoma subjects from Iowa and in 1% of normal-tension glaucoma patients from New York. Affected siblings of the probands AG724 and GFMC524 (AG724.1 and GFMC524.1, respectively) were also shown to carry TBK1 duplications using the quantitative polymerase chain reaction assay. The inheritance of TBK1 copy number variations and normal-tension glaucoma is shown in Figure 2 for these pedigrees. Interestingly, all of the members of 1 pedigree that were diagnosed with normal-tension glaucoma (AG604, AG604.1, and AG604.2) had 2 extra copies of TBK1 (triplication), while previously reported cases had 1 extra copy ( Figure 1 ). All families display an autosomal dominant inheritance pattern of TBK1 copy number variations and normal-tension glaucoma, providing further evidence that these copy number variations are pathogenic. Moreover, these data also suggest that the extra copies of the TBK1 gene are tandem repeats on the same allele, that is, a gene duplication in pedigrees with 1 extra copy of TBK1 and a gene triplication in pedigrees with 2 extra copies.
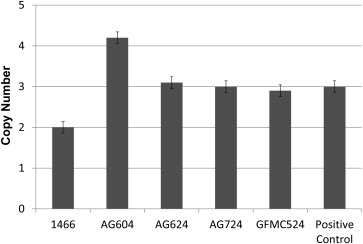
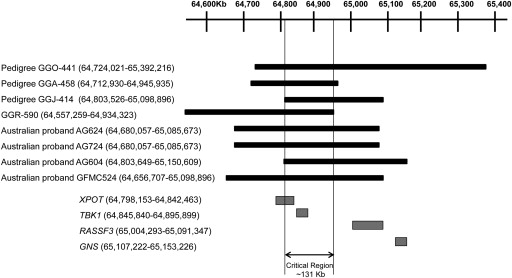
The borders of the copy number variations detected in normal-tension glaucoma probands GFMC524, AG604, AG624, and AG724 were assessed using comparative genomic hybridization ( Figure 3 ). The copy number variations in these Australian normal-tension glaucoma patients are all novel and differ from previously reported copy number variations in the extent of chromosome 12q14 that is involved. Both probands AG624 and AG724 had a duplication, extending from approximately 64,68 Mbp to 65,09 Mbp on chromosome 12. These probands were not known to be related; however, detection of identical copy number variation borders suggested a founder effect. This hypothesis was investigated by comparing haplotypes spanning the TBK1 locus using genotypes obtained from a prior genome-wide association study. These 2 patients were found to share a common haplotype over a greater than 1.4 Mbp segment of chromosome 12q14 (between rs10506464 and rs1909340), which further supports a founder effect in these 2 individuals. A 300 kbp duplication was detected in normal-tension glaucoma proband AG604 that has similar borders as a previously reported copy number variation in a Japanese normal-tension glaucoma patient, GGJ-414 ( Figure 3 ). Genotype data were not available to explore a possible founder effect between these 2 patients. When the copy number variations from the current report and those from prior reports were analyzed, the overlap defined a critical region (∼131 kbp), which harbors the TBK1 gene and part of the XPOT gene ( Figure 3 ).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


