Clinical Trials in Wet Age-Related Macular Degeneration
Stephen A. McNutt MD
Peter K. Kaiser MD
Age-related macular degeneration (AMD) remains the leading cause of blindness in the developed world and is a major cause of blindness worldwide.1 Wet macular degeneration accounts for 10% to 20% of cases of AMD, but makes up 80% to 90% of severe vision loss associated with the disease.2 The management of AMD has drastically changed in the past 10 years. Previously, the available treatments for AMD decreased the incidence of moderate to severe vision loss, but in many cases visual loss still occurred. The Macular Photocoagulation Study (MPS) involved the evaluation of delivery of laser to choroidal neovascularization (CNV) in an effort to halt severe vision loss.3,4,5 Similarly, verteporfin photodynamic therapy (PDT) trials including the Treatment of Age-related macular degeneration with Photodynamic therapy (TAP) study demonstrated a reduced risk for moderate to severe vision loss in certain patients with CNV from AMD, however showed only a minimal proportion of patients gaining vision.6,7,8,9 The pathogenesis of wet AMD includes angiogenesis, the formation of new blood vessels, from existing vasculature. Vascular endothelial growth factor (VEGF) has been shown to play a role in this process of neovascularization in AMD.10 With a better understanding of the relationship of angiogenesis and VEGF in the pathogenesis of wet AMD came more therapies targeted toward VEGF, namely the anti-VEGF antibodies ranibizumab and bevacizumab. These medications have changed the care of patients with AMD and have given them the possibility of regaining lost vision.
This chapter describes the main studies currently relevant for the treatment of wet AMD.
I. ANTI-VEGF ANTIBODY FOR THE TREATMENT OF PREDOMINANTLY CLASSIC CHOROIDAL NEOVASCULARIZATION IN AGE-RELATED MACULAR DEGENERATION (ANCHOR) STUDY
The ANCHOR study was designed to compare the efficacy and safety of intravitreal ranibizumab, a 48-kDa humanized, affinity-matured antibody to VEGF-A isoforms, and PDT with verteporfin in patients with predominantly classic CNV due to AMD. Together, the ANCHOR and MARINA studies lead to a paradigm shift in the management of neovascular AMD.
Study Design
In the ANCHOR study, 423 patients were randomized 1:1:1 to receive 24 monthly intravitreal injections of 0.3 mg or 0.5 mg ranibizumab (Lucentis, Genentech, South San Francisco, CA) with sham PDT or monthly sham injections with standard verteporfin (Visudyne, QLT, Vancouver, BC) PDT. Patients were eligible to receive additional sham or standard PDT treatment every 3 months if they showed leakage from CNV on fluorescein angiography (FA).11
Primary Outcome
Proportion of patients who lost <15 letters at month 12 compared with the baseline in the best-corrected visual acuity (BCVA) score.
Key Secondary Outcomes
Proportion of patients who lost <15 letters at 24 months versus baseline
Mean change in BCVA at 12 and 24 months
Percentage gaining ≥ 15 letters
Change in FA lesion characteristics
Ocular and systemic side effects
Major Inclusion Criteria
Age ≥50
Predominantly classic subfoveal CNV due to AMD
ETDRS BCVA (Snellen equivalent) between 20/40 to 20/320 in the study eye
Lesion eligible for PDT (<9 MPS disc areas [DA])
No prior laser treatment involving the center of the fovea
No prior PDT or experimental treatments for AMD
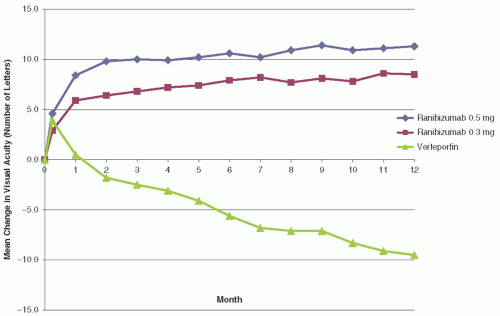 FIGURE 8B.1 Mean change in number of letters read versus month in ranibizumab 0.5 mg, ranibizumab 0.3 mg, and verteporfin treatment groups of the ANCHOR trial. (Reproduced from Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. New Engl J Med. 2006;355:1432-1444, with permission.) |
Results
Efficacy analysis was performed using an intent-to-treat analysis. At month 12, the proportion of patients losing <15 letters was found to be 94.3% in the 0.3 mg ranibizumab group, 96.4% in the 0.5 mg group, and 64.3 % in the PDT group (P < 0.001).11 Figure 8B.1 shows the mean change in visual acuity (VA) (letters) (± standard error) over time in the study through month 12, and clearly shows a significant trend toward gain of vision in the ranibizumab groups versus the PDT group. At 24 months statistical significance remained in this comparison with 90.0% and 89.9% of patients losing <15 letters in the 0.3 mg and 0.5 mg ranibizumab groups, respectively, versus 65.7% in the PDT group.12 A gain of ≥15 letters was seen in 35.7%, 40.3%, and 5.6% of patients in the 0.3 mg ranibizumab, 0.5 mg ranibizumab, and PDT groups at 12 months, respectively (P < 0.001 for ranibizumab
groups vs. sham).11 Similar gains in VA remained at 24 months. The mean change in VA at 12 months was +8.5 and +11.3 letters for the 0.3 mg and 0.5 mg groups, respectively, with a mean loss of -9.5 letters in the PDT group (P < 0.001).11 At 24 months, the mean change in BCVA was +8.1 letters for 0.3 mg ranibizumab, +10.7 letters for 0.5 mg ranibizumab, and -9.8 letters in the PDT group, which remained statistically significant.12 Ranibizumab showed significantly more favorable changes at both 12 and 24 months with respect to total area of CNV lesion, CNV area, and total area of CNV leakage11,12 compared to PDT.
groups vs. sham).11 Similar gains in VA remained at 24 months. The mean change in VA at 12 months was +8.5 and +11.3 letters for the 0.3 mg and 0.5 mg groups, respectively, with a mean loss of -9.5 letters in the PDT group (P < 0.001).11 At 24 months, the mean change in BCVA was +8.1 letters for 0.3 mg ranibizumab, +10.7 letters for 0.5 mg ranibizumab, and -9.8 letters in the PDT group, which remained statistically significant.12 Ranibizumab showed significantly more favorable changes at both 12 and 24 months with respect to total area of CNV lesion, CNV area, and total area of CNV leakage11,12 compared to PDT.
Safety
There were no imbalances in serious or nonserious ocular adverse events (AEs) between the three groups at 24 months.12 Also, no imbalance was seen in serious nonocular AEs between groups. There was no significant difference at 24 months between ranibizumab groups versus PDT when comparing rates of arterial thrombotic events as defined by the Antiplatelet Trialists’ Collaboration.12,13
II. MINIMALLY CLASSIC/OCCULT TRIAL OF THE ANTI-VEGF ANTIBODY RANIBIZUMAB IN THE TREATMENT OF NEOVASCULAR AGE-RELATED MACULAR DEGENERATION (MARINA)
The MARINA study set out to evaluate ranibizumab versus sham injection in patients with minimally classic and occult with no classic CNV due to wet AMD. Taken with the results of the ANCHOR study, the treatment of wet AMD changed direction toward the use of anti-VEGF agents.
Study Design
The MARINA study was a prospective, 2-year, randomized, double-masked, sham-controlled study of the efficacy and safety of the use of ranibizumab in patients with minimally classic and occult with no classic CNV due AMD14; 716 patients were randomized (1:1:1) to receive 24 monthly intravitreal injections with 0.3 mg ranibizumab, 0.5 mg ranibizumab, or sham intravitreal injections in minimally classic or occult with no classic, subfoveal CNV.
Primary Outcome
Proportion of subjects who lost <15 letters at month 12 compared with baseline in their BCVA
Key Secondary Outcomes
Patients who gained 15 or more letters in BCVA from baseline
Mean increase in BCVA from baseline at 12 months
Ocular and systemic side effects
Major Inclusion Criteria
Age ≥50 years
ETDRS BCVA (Snellen equivalent) between 20/40 and 20/320 in the study eye
Subfoveal CNV secondary to AMD
Lesion composition by FA:
Area of CNV must be ≥50% of the total lesion
Minimally classic or occult with no classic CNV
Evidence of presumed recent disease progression as evidenced by new subretinal hemorrhage, recent growth on FA or recent VA loss
Lesion size ≤ 12 DA
Results
Efficacy analysis was performed using an intent-to-treat analysis; 94.5% patients receiving 0.3 mg ranibizumab and 94.6% of those receiving 0.5 mg ranibizumab lost less than 15 ETDRS letters versus 62.6% in the sham group at 12 months (P < 0.001 for both groups vs. sham).14 The significant difference in patients losing <15 letters remained
at 24 months. At 12 months, 24.8% of the patients in the 0.3 mg ranibizumab group and 33.8% in the 0.5 mg group gained ≥15 letters versus 5.0% in the sham group (P < 0.001) with the percentages remaining similar at 24 months.14 The mean change in BCVA from baseline to 12 months was +6.5 letters and +7.2 letters in the 0.3 mg and 0.5 mg ranibizumab groups, respectively versus a loss of 10.4 letters in the sham injection group (P < 0.001).14 Again, the difference between the groups remained at 24 months (Fig. 8B.2).
at 24 months. At 12 months, 24.8% of the patients in the 0.3 mg ranibizumab group and 33.8% in the 0.5 mg group gained ≥15 letters versus 5.0% in the sham group (P < 0.001) with the percentages remaining similar at 24 months.14 The mean change in BCVA from baseline to 12 months was +6.5 letters and +7.2 letters in the 0.3 mg and 0.5 mg ranibizumab groups, respectively versus a loss of 10.4 letters in the sham injection group (P < 0.001).14 Again, the difference between the groups remained at 24 months (Fig. 8B.2).
Safety
There was no significant difference in AEs at 24 months. Seventeen deaths were reported over the 24-month period with similar rates in all groups— approximately 2.5%. Arterial thrombotic events were reported at a rate of 3.8%, 4.6%, and 4.6% in the sham, 0.3 mg ranibizumab, and 0.5 mg ranibizumab groups, respectively.14
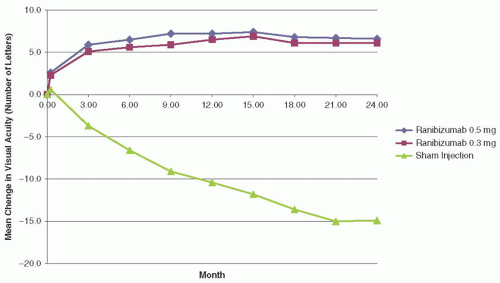 FIGURE 8B.2 Mean change in number of letters read versus month in ranibizumab 0.5 mg, ranibizumab 0.3 mg, and sham injection treatment groups of the MARINA trial. (Reproduced from Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. New Engl J Med. 2006;355:1419-1431, with permission.) |
III. A PHASE IIIB, MULTICENTER, RANDOMIZED, DOUBLE-MASKED, SHAM INJECTION-CONTROLLED STUDY OF THE EFFICACY AND SAFETY OF RANIBIZUMAB IN SUBJECTS WITH SUBFOVEAL CHOROIDAL NEOVASCULARIZATION WITH OR WITHOUT CLASSIC CNV SECONDARY TO AGE-RELATED MACULAR DEGENERATION STUDY (PIER) STUDY
The PIER study set out to study an alternate dosing regimen of ranibizumab to the monthly treatment protocol studied in the MARINA and ANCHOR studies.
Study Design
This was a 2-year, Phase IIIb, multicenter, double-masked, sham injection-controlled evaluation of the efficacy and safety of ranibizumab in patients with subfoveal CNV due to AMD.15
Patients included those with and without classic CNV. The study evaluated quarterly dosing, an alternative and less frequent regimen than the monthly ranibizumab dosing tested in MARINA and ANCHOR; 184 patients were randomized to one of three treatment groups in a 1:1:1 fashion. Treatment groups included those receiving 0.3 mg ranibizumab, 0.5 mg ranibizumab, and sham injections. Patients in the ranibizumab groups received injections at day zero, month 1, and month 2 with subsequent doses every 3 months thereafter irrespective of clinical exam or findings. After 12 months of evaluation, patients in the sham injection group were allowed to “crossover” to receive 0.5 mg ranibizumab every 3 months. It was later deemed necessary to offer monthly injections with 0.5 mg ranibizumab for the duration of the 2-year study “roll over” patients.16
Patients included those with and without classic CNV. The study evaluated quarterly dosing, an alternative and less frequent regimen than the monthly ranibizumab dosing tested in MARINA and ANCHOR; 184 patients were randomized to one of three treatment groups in a 1:1:1 fashion. Treatment groups included those receiving 0.3 mg ranibizumab, 0.5 mg ranibizumab, and sham injections. Patients in the ranibizumab groups received injections at day zero, month 1, and month 2 with subsequent doses every 3 months thereafter irrespective of clinical exam or findings. After 12 months of evaluation, patients in the sham injection group were allowed to “crossover” to receive 0.5 mg ranibizumab every 3 months. It was later deemed necessary to offer monthly injections with 0.5 mg ranibizumab for the duration of the 2-year study “roll over” patients.16
Major Inclusion Criteria
Age ≥50 years
Primary or recurrent subfoveal CNV due to AMD with ≥50% of the lesion composed of active CNV
Total AMD lesion size ≤ 12 DA
BCVA of 20/40 to 20/320 Snellen equivalent as per ETDRS charts
Minimally classic and occult lesions were eligible if they met any of the following:
≥10% increase in lesion size on FA measured ≤ 1 month versus ≤ 6 months prior to day zero
>1 Snellen VA line loss, or equivalent within 6 months prior to day zero
CNV-associated hemorrhages ≤ 1 month prior to day zero
Exclusion Criteria
Any prior treatment with verteporfin PDT, external-beam radiation, transpupillary thermotherapy, or subfoveal laser photocoagulation
Juxtafoveal or extrafoveal laser photocoagulation ≤ one month prior to day zero
Permanent structural damage to the central fovea
Subretinal hemorrhage involving the fovea if ≥1 DA or ≥50% total lesion area
Treatment of either eye in a previous anti-angiogenic treatment trial
PDT in the nonstudy eye ≤ 7 days prior to day zero
Primary Outcome
Mean change in VA from baseline to 12 months
Secondary Outcomes
Mean change in VA from baseline to 24 months
Proportion of patients who lost <15 VA letters from baseline
Proportion of patients who gained ≥15 VA letters from baseline
Proportion of patients with Snellen equivalent VA of 20/200 or less
Mean change from baseline in total area of CNV and total area of leakage from CNV
Results
All randomized patients were evaluated with an intent-to-treat analysis with last observation carried forward; 85% or more patients in the ranibizumab groups received all scheduled injections and 27% of patients in the sham group discontinued treatment prior to 12 months. The mean change in VA from baseline was -16.3 letters in the sham group versus -1.6 letters and -0.2 letters in the 0.3 mg ranibizumab and 0.5 mg ranibizumab groups, respectively (P = 0.0001 and P < 0.0001, respectively) at 12 months. VA was compared at 3 and 12 months to evaluate quarterly “maintenance” dosing. Both ranibizumab groups lost an average of -4.5 letters between month 3 and 12 with both declines considered significant as none of the 95% confidence intervals included zero.
The proportion of patients losing <15 Snellen letters was significantly lower in the ranibizumab groups versus the sham groups with 83.3% and 90.2% in the 0.3 mg and 0.5 mg groups losing < 15 letters, respectively, and 49.2% in the sham group (P < 0.0001 for both groups vs. sham). There was no significant difference in the proportion of patients gaining at least 15 letters versus sham. There
were significantly fewer patients in the ranibizumab groups with ≤ 20/200 Snellen VA versus sham. Both ranibizumab groups showed a significant reduction in growth of CNV and in total area of leakage from CNV.
were significantly fewer patients in the ranibizumab groups with ≤ 20/200 Snellen VA versus sham. Both ranibizumab groups showed a significant reduction in growth of CNV and in total area of leakage from CNV.
At 24 months, mean loss from baseline VA was -21.4, -2.2, and -2.3 letters in the sham, 0.3 mg, and 0.5 mg ranibizumab groups, respectively (P < 0.0001 for both ranibizumab vs. sham).16 The total area of CNV remained significantly different at 24 months with an increase in 1.9 disc areas (DA) in the sham group and 0.29 DA in the 0.3 mg group (P = 0.0015) and 0.64 DA in the 0.5 mg group (P = 0.0021).
Safety
There were no cases of endophthalmitis during the 2 years. Rates of arteriothrombotic events (ATE) were zero in all groups at 1 year. Rates of ATEs at 2 years were reported at 1.6% in the postcrossover from sham group, 1.7% in the postcrossover 0.3 mg group, and 0% in the postcrossover 0.5 mg group.
IV. A PHASE IIIB, OPEN-LABEL, MULTICENTER 12-MONTH STUDY TO EVALUATE THE SAFETY, TOLERABILITY, AND EFFICACY OF RANIBIZUMAB (0.3 MG AND/OR 0.5 MG) IN PATIENTS WITH SUBFOVEAL CHOROIDAL NEOVASCULARIZATION SECONDARY TO AGE-RELATED MACULAR DEGENERATION: THE SUSTAIN STUDY
SUSTAIN looked to evaluate PRN dosing of ranibizumab based on defined retreatment criteria including change in vision and OCT central retinal thickness (CRT).
Study Design
This was a 12-month, phase III, multicenter, open-label, single-arm study evaluating the safety and efficacy of intravitreal ranibizumab in patients with CNV secondary to AMD.17 Patients received three monthly doses of 0.3 mg ranibizumab intravitreally followed by monthly evaluations and intravitreal injections on an as-needed basis (PRN) as defined by prespecified criteria. As-needed injections were given in the setting of VA loss of >5 letters or an increase in CRT of >100 µm. After January 2007, patients received 0.5 mg ranibizumab as opposed to 0.3 mg.
Inclusion Criteria
Age ≥ 50 years
Diagnosis of active primary or recurrent subfoveal CNV secondary to AMD
Total area of CNV ≥ 50% of total lesion area
Total lesion area ≤ 12 DA
BCVA between 73 and 24 ETDRS letters (˜20/40 to 20/320 Snellen equivalent)
Primary Outcome
Incidence and severity of ocular AEs over 12 months
Key Secondary Outcomes
Mean change from baseline to 3 and 12 months in BCVA and CRT
Total treatments
Results
455 of the initial 513 enrolled patients completed 12 months of treatment; 48.5% of patients experienced at least one ocular AE, including reduced VA, retinal hemorrhage, increased intraocular pressure, and conjunctival hemorrhage17; 3.7% of patients experienced arterial thrombotic events, with 1% experiencing a transient ischemic attack, cerebral infarction, or cerebrovascular accident.17 There were seven deaths during the 12 months of treatment with one death caused by an event thought to be associated with ranibizumab.17
Mean change in BCVA from baseline was +5.8 and +3.6 letters at months 3 and 12, respectively.17 A steady increase in BCVA change was observed over the first 3 months with a decrease from month 3 to 6 and a stable period from month 6 to 1217; 96.7% and 92.5%
of patients lost <15 letters at months 3 and 12, respectively. Change in CRT was -101.1 µm at month 3 from baseline with a change of -91.5 seen at month 12 versus baseline.17 The mean number of injections over 12 months was 5.6 when including the three initial injections.17
of patients lost <15 letters at months 3 and 12, respectively. Change in CRT was -101.1 µm at month 3 from baseline with a change of -91.5 seen at month 12 versus baseline.17 The mean number of injections over 12 months was 5.6 when including the three initial injections.17
Although SUSTAIN is a single-arm, unmasked study, the authors contend that the trial is comparable to MARINA and ANCHOR in that patient CNV characteristics and study designs were similar. The authors note ˜80% efficacy of PRN injections versus monthly injections when comparing treatments to controls.
V. EFFICACY AND SAFETY OF MONTHLY VERSUS QUARTERLY RANIBIZUMAB TREATMENT IN NEOVASCULAR AGE-RELATED MACULAR DEGENERATION: THE EXCITE STUDY
The EXCITE study was designed to evaluate the monthly dosing regimen of ranibizumab as in the MARINA and ANCHOR trials versus quarterly dosing as performed in the PIER study.
Study Design
This was a 1-year, randomized, multicenter, active-controlled, Phase IIIb study evaluating efficacy and safety of monthly versus quarterly dosing of ranibizumab in patients with subfoveal CNV secondary to AMD.18 Patients were randomly assigned in a 1:1:1 fashion to one of three treatment groups. Patients received either three consecutive monthly intravitreal loading doses of 0.3 mg ranibizumab (arm A) or 0.5 mg ranibizumab (arm B) followed by quarterly dosing (every three months) or 0.3 mg ranibizumab monthly for the duration of the study (arm C). Patients in arm A and B received sham injections monthly when not receiving ranibizumab to maintain masking.
Inclusion Criteria
Age ≥ 50 years
Total area of CNV ≥ 50% of total lesion area
Total lesion area ≤12 DA for minimally classic or occult with no classic, or ≤ 9 DA for predominantly classic lesions
BCVA between 73 and 24 letters (approximately 20/40 to 20/320 Snellen equivalent)
Primary Outcome
Mean change in BCVA from baseline to month 12
Results
353 of 482 screened patients were randomized to one of the three treatment arms. Mean BCVA increased from baseline +4.9, +3.8, and +8.3 letters in the 0.3 mg quarterly, 0.5 mg quarterly, and 0.3 mg monthly groups, respectively, at 12 months in the per-protocol analysis.18 Mean BCVA increase in this analysis over the first 3 months was +6.8, +6.6, and +7.5 letters in the 0.3 mg quarterly, 0.5 mg quarterly, and 0.3 mg monthly groups respectively with quarterly groups subsequently losing acuity after the baseline monthly loading doses.18 The intent-to-treat analysis showed similar results as the per-protocol analysis. Both analyses show an initial and similar increase in VA over the first 3 months (with monthly dosing in all arms) with subsequent decreases in mean BCVA in the quarterly arms versus monthly arm at 12 months. Noninferiority could not be shown for the quarterly versus monthly treatment arms.18
VI. THE PHASE III, DOUBLE-MASKED, MULTICENTER, RANDOMIZED, ACTIVE TREATMENT-CONTROLLED STUDY OF THE EFFICACY AND SAFETY OF 0.5 MG AND 2.0 MG RANIBIZUMAB ADMINISTERED MONTHLY OR ON AN AS-NEEDED BASIS (PRN) IN PATIENTS WITH SUBFOVEAL NEOVASCULAR AGE-RELATED DEGENERATION: THE HARBOR STUDY
The HARBOR study set out to evaluate a higher dose of ranibizumab in a monthly and
a PRN dosing regimen versus the standard dose in patients with wet AMD.
a PRN dosing regimen versus the standard dose in patients with wet AMD.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


