Purpose
To review what progress has been made towards the application of ocular gene therapy to prevent progressive vision loss in patients affected by choroideremia.
Design
A Perspective based on the collective opinions of researchers and clinicians actively engaged in vision research on choroideremia and a review of current literature.
Methods
Researchers from Europe, Canada, Australia, and the United States were convened to the first International Choroideremia Research Symposium held in Sommières, France in September 2011. Attendees shared their collective understanding of the pathophysiology of choroideremia and current trends in the development of treatments, with an emphasis on the potential of gene therapy as an achievable approach. Supplemental perspectives are provided along with an update of progress made since the meeting.
Results
The complexity of treating a retinal disease such as choroideremia that affects multiple tissue layers has been brought to light. The genetic basis of choroideremia must be thoroughly deciphered and appropriate clinical tests selected to follow disease progression and evaluate the efficiency of treatments.
Conclusions
Whereas the time frame for the development of therapies for some retinal dystrophies may be in the years hence, gene therapy trials for choroideremia have started in the United Kingdom and results are pending. These first trials may help resolve the remaining issues associated with the treatment of this disease.
Choroideremia is an X-linked chorioretinal dystrophy, primarily affecting male subjects, with an estimated prevalence of 1 in 50 000. It is characterized by progressive atrophy of the choroid, retinal pigment epithelium (RPE), and photoreceptors. The gene associated with choroideremia ( CHM ; Xq21.2) encodes a ubiquitously expressed protein (Rab escort protein [REP-1]) that enables post-translational isoprenyl modification of Rab proteins (see glossary of potentially unfamiliar terms, available at AJO.com ). Mutations in the CHM gene cause defects in intracellular membrane traffic pathways, including melanosome movement and phagosome processing. There are some variations in severity of disease, including intrafamilial variability, which may be partly explained by differences in the altered levels of trafficking in the cells of these patients. Choroideremia is in most cases nonsyndromic, but has been reported in association with mental retardation and deafness as a contiguous gene syndrome.
Methods
The pathogenesis of choroideremia within the eye remains open to investigation and discussion. To evaluate the complexities of the disease and elaborate the most promising strategy for its treatment, the first International Choroideremia Research Symposium was held in Sommières, France in September 2011. The aim of this symposium was to bring together choroideremia experts from around the world to share information and work towards a therapy. The symposium culminated in an exchange with patients to address their questions.
Results
This Perspective provides expert opinion, derived from the symposium and current literature, on the pathophysiology of choroideremia and trends in treatment, with an emphasis on the potential of gene therapy.
Clinical Aspects
Boys affected with choroideremia complain of difficulty seeing at night in the first decade of life and become aware of loss of peripheral vision by their teens and twenties. By the fourth decade, male individuals with choroideremia have a significantly constricted peripheral visual field. Suitable imaging techniques are required to observe clinical manifestations and choroideremia disease progression, and may have the potential to monitor treatment efficacy. Firstly, optical coherence tomographic (OCT) imaging provides high-resolution in vivo imaging to define progressive retinal changes and reveals areas of hypopigmentation of the RPE and thinning throughout the fundus in the early phase of choroideremia. Retinal thickening occurs subsequently with normal laminae, possibly attributable to Müller cell activation and hypertrophy creating interlaminar bridges. Later phases show typical shortening of the inner and outer segments, reduced thickness of the outer nuclear layer, and depigmentation of the RPE. Areas of chorioretinal atrophy become evident over time with loss of choriocapillaris, exposure of choroidal vessels, and loss of the RPE beyond the macula ( Figure ).
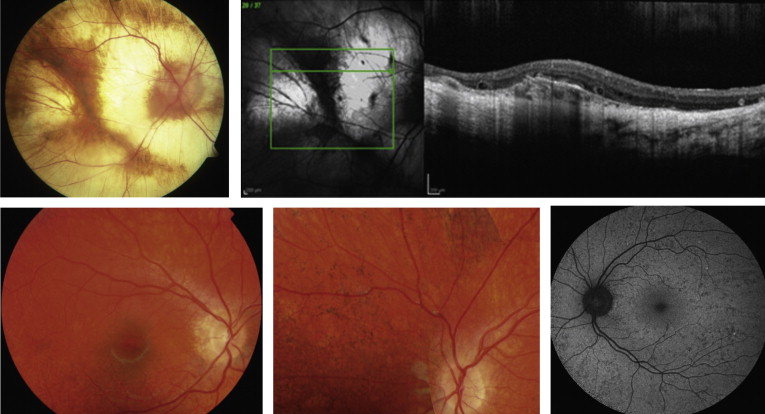
Secondly, fundus autofluorescence (FAF) provides a noninvasive approach to imaging of the RPE. In male subjects affected with choroideremia, FAF allows specific delineation of the remaining RPE and observations of the progressive shrinkage of the central RPE island at rates that can be calculated for individual patients. The rate of loss is greater in younger than older patients. By combining FAF with OCT, the loss of both RPE and photoreceptors can be followed. The observation of early photoreceptor loss in the vicinity of disappearing RPE suggests a concomitant pathologic process occurring in both photoreceptors and RPE cells.
With a milder phenotype, female carriers can be difficult to recognize in childhood. However, detection is aided by FAF imaging, which reveals a pattern of mixed hypoautofluorescent spots, which could correspond to atrophy of the RPE, and hyperautofluorescent spots, which could signal dysfunction of photoreceptors and/or accumulation of lipofuscin in RPE cells ( Figure ). Occasionally, female carriers are severely affected and exhibit a phenotype not unlike male subjects with choroideremia. Although lyonization has traditionally explained the clinical findings in affected female subjects, a plausible explanation may also be insufficiency of REP activity. By inference, the combination of lyonization and the carrier state may deplete tissues such as the RPE of REP-1 and manifest as disordered cellular function. Symptomatic choroideremia carriers may result from skewed lyonization or variability in X inactivation.
Further dissection of the carrier phenotype in choroideremia can be performed by combining fundus-controlled perimetry, multifocal electroretinography (mfERG), 2-color threshold perimetry, spectral-domain OCT, and FAF. Two-color threshold perimetry of choroideremia carriers demonstrates that most have a more pronounced loss of rods, but some have a more prominent loss of cones. Whether cone loss is secondary to the RPE impairment or to a primary cone dysfunction is not known. mfERG superimposed on FAF images shows no strict correlations between abnormal FAF spots and changes in mfERG responses.
Genetics
The CHM gene consists of 15 exons, spans 186 kilobases (kb), and encodes a polypeptide of 653 amino acids. Mutations of the CHM gene that lead to premature termination of translation of REP-1 or the absence of REP-1 include sizeable deletions (20%), point mutations (60%), intronic mutations (10%) and unknown causes (10%) Using mRNA analysis, a deep intronic mutation has been detected in intron 4, resulting in a pseudoexon containing a stop codon, and in intron 12, leading to exon skipping. An L1-retrotransposon insertion that occurred de novo in a choroideremia patient has also been identified. The identification of CHM mutations is important for genetic counseling, accurate clinical management, and on-going therapies.
An autosomal homologue of CHM designated CHML , for choroideremia-like, has also been identified. The gene product of CHML is REP-2, which has an overlapping but not identical function to REP-1. It is believed that REP-2 compensates for a deficiency of REP-1 in all human tissues, except the eye.
Recently, several databases have been developed for CHM genetic variations, including LOVD-CHM (Leiden Open Variation Database; http://www.lovd.nl/CHM ), an open-source tool that provides a complete and accurate repository of published mutations occurring in CHM . Currently, it contains 127 published CHM variants, 259 entries, and 36 PubMed references. A second LOVD-CHM database dedicated to published mutations is available at http://ngrl.manchester.ac.uk/LOVDv.2.0/home.php?select_db=CHM .
Gene Therapy
The field of gene therapy for blinding disorders of the retina is at a turning point because of recent advances in proof-of-concept studies, improvements in vectors and technology, and the first-in-human experiences. The summit was attained in 2008 by the first phase I/II clinical trials for an autosomal recessive, congenital form of severe blindness, Leber congenital amaurosis (LCA), caused by mutations in RPE65 , a gene specifically expressed in the RPE. The encouraging results and safety data collected from these and additional trials have paved the way to retinal gene transfer for other monogenic diseases, such as choroideremia.
Many of the difficulties associated with designing gene therapy for choroideremia surround the choice of vector. The first issue is that the choroid is the most active blood supply in the body. Thus, any vector administered to the choroid will be flushed away instantly. It is not possible to stop blood flow for a limited time without damage; therefore, it is important to resolve the enigma of whether choroidal atrophy (which appears early in disease pathogenesis) is secondary to RPE degeneration. The second consideration is which cell types to target. The consensus is that both RPE and photoreceptors should be targeted. A third issue is the promoter to use. The clinical RPE65 trials with a cell-specific promoter have shown the least amelioration in patients, suggesting that a strong promoter is required for sufficient expression of exogenous protein. This is reassuring, as a ubiquitous promoter is likely needed for choroideremia, since 2 tissue types must be targeted. The final consideration is the intellectual property associated with the chosen serotype. This can cause a significant hurdle by blocking the administration of the final drug to patients.
Viral vectors
Various vectors, both viral and nonviral, have been developed to facilitate the entry of DNA to cells. Viral systems are more advanced and have already been tested in the clinic. To generate a safe viral system, the replicative functions of a virus must be eliminated to produce a vector in which the DNA of interest can be encapsulated. A viral vector should be able to safely and stably deliver its cargo into the cell without eliciting an immune response. One of the first vectors to be tested in the retina was derived from human adenoviruses (Ad). However, first-generation Ad vectors are relatively immunogenic because of the presence of some viral genes, thus limiting their use. This characteristic led to the development of a “gutted” Ad vector system devoid of all viral genes. This vector has a large cargo capacity and is less immunogenic; however, gutted adenovirus vectors are difficult to prepare and have not been validated in the clinic.
In 1996, the first adeno-associated viral (AAV) vector was tested in the retina. AAVs are small (20 nm in diameter), nonpathogenic viruses. AAV vectors are “gutted” in that only the inverted terminal repeats are conserved from the viral genome, between which the transgene of interest and its regulatory elements are inserted. By manipulating the AAV capsid, it is possible to alter both its tropism and the lag time prior to DNA expression, thus widening its potential applications. In 2001, development of the lentiviral vector made it possible to target larger genes because of a larger cloning capacity (≥7.5 kb) than AAV vectors (∼4.7 kb). However, lentiviral vectors are less efficient for targeting photoreceptors than AAV. Regardless, a lentiviral vector is currently being tested in a clinical trial for cone-rod dystrophy forms of Stargardt disease ( ClinicalTrials.gov identifier: NCT01367444 ) attributable to mutations in the ABCA4 gene.
To date, in vivo data have not been reported with AAV vectors in choroideremia, but in vivo gene transfer studies have been performed using HIV-based lentiviral vectors expressing CHM under control of the elongation factor-1 alpha promoter. This vector was administered by subretinal delivery into REP-1-deficient mice, and a specific transduction of the RPE was detected. Photoreceptor cells were not transduced, although some Müller cells at the site of injection expressed the transgene, which was likely attributable to a destabilization of the retinal structure. A restoration of function was detected by a reduction in the number of unprenylated Rabs. Transgene expression was observed for 6 months, and no toxic effects were seen. However, the lack of photoreceptor transduction suggests that lentiviral-based vectors will be of limited use for choroideremia.
Nonviral vectors
Despite the promise of viral-mediated gene therapy for the eye, research continues to explore other, potentially safer, nonviral alternatives. In addition to safety issues, plasmid-based vectors can have unlimited cloning capacity, which will allow the introduction of introns as well as exons of a gene in addition to promoter and other regulatory sequences. Such a cassette can contribute to natural cell-specific control as well as allowing sustained expression in the absence of integration. The unlimited cloning capacity is particularly attractive considering the small capacity of AAV vectors, which in the case of some larger genes precludes even the cloning of a complete cDNA.
A novel plasmid vector employing a scaffold/matrix attachment region (S/MAR) has been developed. S/MARs are DNA-binding motifs found throughout normal chromosomes that anchor the chromatin to the nuclear scaffold. When included into a plasmid vector, S/MARs confer episomal maintenance, prevent epigenetic silencing, and mediate extrachromosomal replication. Moreover, S/MAR vector technology is applicable in vitro (as well as in vivo and ex vivo), in contrast to certain AAV vectors. To test the potential of using this technology in choroideremia, a S/MAR vector containing the CHM gene under control of the CAG promoter (a combination of the chicken beta-actin promoter and the cytomegalovirus early enhancer element) was generated and administered subretinally in mice. REP-1 expression was seen in the RPE and no toxicity or damage was observed.
Choroideremia and gene therapy: A surgical challenge?
Gene therapy requires the injection of vectors close to the damaged tissue. In choroideremia, this cannot be easily achieved using conventional subretinal injection, as atrophy of both the RPE and photoreceptors weakens the link between the 2 cell types and increases the possibility of retinal detachment, and reduces contrast and hence visibility. In addition, because of atrophy of the choroid, retinal fluid absorption takes longer than in other inherited retinal diseases. A potentially better alternative to subretinal injection may be suprachoroidal drug delivery. A suprachoroidal catheter containing a light fiber is inserted tangentially to the sclera and the light is used to guide the end of the probe to the front of the macula. It is a relatively simple surgery and a promising technique. However possible complications, notably those arising on an atrophic retina, have not yet been established.
Clinical trials
The first gene replacement clinical trial for choroideremia was conducted in the United Kingdom in October 2011 ( ClinicalTrials.gov Identifier: NCT01461213 ). An AAV2/2 vector was used to vehicle the CHM gene by subretinal injection (with detachment of the central macula) into 2 cohorts of 3 patients with, to date, no adverse side effects. More clinical trials are predicted to begin in Canada, the United States, and France over the next few years.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


