The pituitary gland functions prominently in the control of most endocrine systems in the body. Diverse processes such as metabolism, growth, reproduction, and water balance are tightly regulated by the pituitary in conjunction with the hypothalamus and various downstream endocrine organs. Benign tumors of the pituitary gland are the primary cause of pituitary pathology and can result in inappropriate secretion of pituitary hormones or loss of pituitary function. First-line management of clinically significant tumors often involves surgical resection. Understanding of normal pituitary physiology and basic testing strategies to assess for pituitary dysfunction should be familiar to any skull base surgeon.
Key points
- •
Benign pituitary tumors are the most common cause of clinically relevant pituitary disease.
- •
The most significant anatomic consequence of an enlarging pituitary tumor is compression of the optic chiasm resulting in visual field loss.
- •
Evaluation of incidentally found pituitary tumors should include assessment for oversecretion of prolactin and growth hormone; assessment for Cushing disease should be undertaken if the clinical presentation is suspicious.
- •
First-line treatment of prolactinoma is medical therapy, not surgical resection.
- •
Perioperative assessment for adrenal insufficiency and hypothyroidism should be performed on all patients with a pituitary tumor as deficiencies of these hormones can lead to perioperative complications in the short term and can result in death in cases of long-term deficiency.
Introduction
The pituitary gland functions as a master regulator of numerous physiologic processes via its control of downstream endocrine glands. The pituitary controls the adrenals, gonads, and thyroid gland via secretion of specific regulating hormones into the systemic circulation. Growth and water balance also represent key physiologic processes directly influenced by the release of pituitary hormones. Pathology of the pituitary gland is primarily caused by benign pituitary tumors (adenomas) that can result in hormonal hypersecretion, impairment of normal pituitary function, or mass effect on surrounding structures. Other sellar lesions, such as cysts or nonadenomatous tumors, can also cause mass effect or impairment of pituitary function. Pituitary diseases cause varied clinical presentations which manifest across all medical disciplines; a basic understanding of pituitary anatomy, physiology, and the process for evaluating pituitary dysfunction is critical for any skull base surgeon.
This article reviews relevant anatomic relationships between the pituitary gland and surrounding structures while also examining the regulation and downstream action of pituitary hormones. Dysfunction of the pituitary gland which typically results from benign pituitary tumors and the clinical manifestations of these tumors, ranging from mass effects on surrounding structures to hormonal hypersecretion or hyposecretion, is reviewed. The biochemical evaluation of suspected pituitary function abnormalities is then presented and suggested testing strategies are outlined for various disease states.
Introduction
The pituitary gland functions as a master regulator of numerous physiologic processes via its control of downstream endocrine glands. The pituitary controls the adrenals, gonads, and thyroid gland via secretion of specific regulating hormones into the systemic circulation. Growth and water balance also represent key physiologic processes directly influenced by the release of pituitary hormones. Pathology of the pituitary gland is primarily caused by benign pituitary tumors (adenomas) that can result in hormonal hypersecretion, impairment of normal pituitary function, or mass effect on surrounding structures. Other sellar lesions, such as cysts or nonadenomatous tumors, can also cause mass effect or impairment of pituitary function. Pituitary diseases cause varied clinical presentations which manifest across all medical disciplines; a basic understanding of pituitary anatomy, physiology, and the process for evaluating pituitary dysfunction is critical for any skull base surgeon.
This article reviews relevant anatomic relationships between the pituitary gland and surrounding structures while also examining the regulation and downstream action of pituitary hormones. Dysfunction of the pituitary gland which typically results from benign pituitary tumors and the clinical manifestations of these tumors, ranging from mass effects on surrounding structures to hormonal hypersecretion or hyposecretion, is reviewed. The biochemical evaluation of suspected pituitary function abnormalities is then presented and suggested testing strategies are outlined for various disease states.
Basic anatomy
The pituitary gland lies in the sella turcica at the base of the skull and consists of 2 lobes (anterior and posterior) which arise from different embryologic origins. The anterior lobe originates from the oropharynx (Rathke’s pouch) and grows upward during development where it meets tissue of neural origin growing inferiorly; this neural tissue forms the posterior lobe. On completion of development, the posterior lobe contains nerve endings of neurons originating in the hypothalamus. The pituitary stalk connects the hypothalamus to the pituitary gland and serves to transmit the axons of the hypothalamus to the posterior lobe; it also transmits regulatory hormones from the hypothalamus to the anterior lobe via a portal system. The anterior lobe is larger than the posterior lobe and represents approximately two-thirds of total pituitary volume.
Pituitary tumors may cause symptoms as a result of a mass effect on surrounding structures, hence an understanding of clinically relevant surrounding structures is paramount. Dura surrounds the pituitary gland and continues superiorly to form a roof over the sella (diaphragma sella). The cavernous sinuses contain cranial nerves (CN) 3, 4, 6, and the first and second branches of the fifth cranial nerve (V1 and V2). The optic chiasm rests approximately 5 to 10 mm above the diaphragma sella. Enlargement of the gland caused by inflammation, infiltration, or tumor growth can result in compression of these structures leading to bitemporal hemianopsia (from compression of the optic chiasm), extraocular muscle dysfunction (from palsies of CN 3,4, or 6), or ipsilateral facial pain (from involvement of the V1 and V2 branches). Pituitary adenomas are classified based on their functional status (nonfunctioning vs hormonally active) and size; tumors up to 1 cm in diameter are microadenomas, whereas tumors greater than 1 cm are macroadenomas.
Physiology
The anterior pituitary gland consists of 5 separate cell types each of which secrete different hormones. Each cell type originates from a common progenitor cell in response to expression of specific transcription factors; deficiencies in these transcription factors result in genetic syndromes of hypopituitarism caused by a lack of particular anterior pituitary cell types. Secretion of hormones from the anterior pituitary is a tightly regulated process influenced by physiologic stimuli and feedback control from downstream effector organs. In general, releasing hormones from the hypothalamus travel to the pituitary gland via the portal hypophysial circulation; these releasing hormones trigger the release of anterior pituitary hormones which then act on their target organ to exert biologic effects (eg, lactation in the breast in response to prolactin) or to induce hormone secretion from the target organ (eg, secretion of cortisol from the adrenal gland in response to adrenocorticotropic hormone, ACTH). The physiologic functions of the various endocrine axes controlled by the anterior pituitary are listed in Table 1 . It should be noted that growth hormone (GH) can also act directly on target tissues to regulate metabolism and body composition independent of its effects on increasing insulin-like growth factor 1 (IGF-1) secretion from the liver.
| Hormone | Target | Effects on Target | Downstream Effects |
|---|---|---|---|
| ACTH | Adrenal gland | Induction of cortisol secretion | Metabolism regulation; resistance to physiologic stress; maintenance of vascular tone |
| GH | Liver, skeleton, soft tissues | IGF-1 secretion (from liver); growth and regulation of nutrient metabolism | IGF-1 is primary mediator of growth |
| FSH and LH | Testes/ovaries | Secretion of testosterone or estrogen/progesterone | Maintenance of fertility, lean body mass, and bone density |
| PRL | Breast | Lactation | — |
| TSH | Thyroid gland | Induction of thyroid hormone (T4) secretion | Metabolism regulation |
Negative regulation of most anterior pituitary hormones occurs via feedback of target organ hormones on the hypothalamus and/or the anterior pituitary; an illustrative overview is provided in Fig. 1 . Negative control of thyroid hormone (free T4, FT4), cortisol, GH, and the reproductive hormones is achieved via this paradigm. It should be noted that negative regulation of GH is also mediated by somatostatin, a hormone which increases in response to GH and IGF-1. This physiology has been exploited through the use of synthetic somatostatin analogues to treat GH-producing tumors.
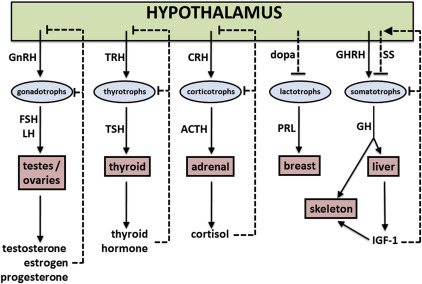
Regulation of prolactin differs from other anterior pituitary hormones in that prolactin secretion is tonically inhibited by dopamine originating from hypothalamic neurons. Physiologic triggers of prolactin secretion (sleep, stress, suckling, pregnancy) decrease hypothalamic secretion of dopamine, thereby allowing lactotrophs to secrete prolactin. Pharmacologic blockade of lactotroph dopamine receptors (eg, by antipsychotics) also leads to increased prolactin secretion.
Overview of pituitary dysfunction
Abnormalities of pituitary function can occur as a result of inappropriate/unregulated hormonal secretion or deficient hormonal secretion. Hormonal hypersecretion often results in physical and biochemical findings characteristic of a particular hypersecretion syndrome (eg, Cushing’s disease, acromegaly). Hypopituitarism can present along a clinical spectrum that depends on the degree of deficiency of the hormone in question and whether this deficiency occurs in isolation or in combination with other pituitary hormones (panhypopituitarism).
Several pituitary hypersecretion syndromes cause significant morbidity and shorten lifespan ; hence early diagnosis and appropriate management are critical. The various hypersecretion syndromes are summarized in Table 2 . Of note, functional gonadotropin tumors (resulting in hypergonadism) are not discussed in this article as they are exceedingly rare. Prolactinoma represents the most common hypersecretion syndrome followed by acromegaly, Cushing’s disease, and central hyperthyroidism caused by a thyrotropin-stimulating hormone (TSH) producing adenoma (“TSH-oma”), respectively. In general, the preferred initial management of hypersecreting pituitary tumors is surgical removal, with the exception of prolactinoma which can be treated medically in most cases. Hence, it is critical for the skull base surgeon to exclude prolactinoma before resecting a pituitary tumor.
| Condition | Presentation | Important Considerations |
|---|---|---|
| Prolactinoma | Galactorrhea, hypogonadism | Medication history critical; surgery reserved for patients who fail medical therapy |
| Acromegaly (↑GH) | Soft tissue overgrowth, hyperhidrosis, HTN, DM2 | IGF-1 is preferred diagnostic test |
| Cushing disease (↑ACTH) | Centripetal obesity, pigmented striae, insomnia, mood lability, skin thinning, proximal muscle weakness, HTN, DM2, hypogonadism | Presentation can be subtle and diagnosis challenging |
| TSH secreting adenoma (TSH-oma) | Weight loss, heat intolerance, hyperdefecation | Diagnosis suggested by hyperthyroid symptoms with ↑FT4 and ↑ (or inappropriately normal) TSH |
In all cases of suspected pituitary dysfunction (either hypopituitarism or hypersecretion syndromes) biochemical testing represents the initial diagnostic step. Once underproduction or inappropriate overproduction of pituitary hormones is confirmed then MRI of the sella (if not already done) should be performed to assess for a pituitary adenoma (the most common cause of pituitary dysfunction). Dedicated pituitary protocol MRI with 1.5-mm cuts provide clear definition of hypothalamic/pituitary anatomy; the addition of contrast is recommended and enhances detection of small pituitary tumors.
Diagnosis of common hypersecretion syndromes
Acromegaly
Acromegaly (GH excess) results in significant morbidity and increases mortality ; however, the diagnosis is often delayed by years due to insidious onset. Assessment for GH oversecretion should be considered in patients with suggestive clinical features and is mandatory in any patient with a pituitary adenoma. Measurement of IGF-1, a growth factor produced in the liver in response to GH stimulation, is the recommended screening test for GH oversecretion. IGF-1 functions as an integrated measure of GH secretion over time and a normal IGF-1 level essentially excludes the diagnosis of acromegaly. Whereas measured serum GH levels are often increased in acromegaly, the pulsatile nature of GH secretion makes a random GH an unreliable means of diagnosing acromegaly.
Once an increase in IGF-1 is demonstrated the diagnosis should then be confirmed with dynamic testing involving oral glucose loading (oral glucose tolerance test), typically done under the supervision of an endocrinologist. This test takes advantage of the ability of hyperglycemia to suppress GH secretion in normal individuals; lack of GH suppression is characteristic of acromegaly.
Cushing’s Disease
The proper diagnosis of Cushing’s syndrome (hypercortisolism) can be a challenging endeavor requiring repeated assessment of endocrine function. Most cases of Cushing syndrome are caused by excess ACTH secretion from a pituitary adenoma (Cushing disease) ; the primary issue likely to be encountered by skull base surgeons surrounds the question of whether a patient with a known pituitary adenoma (usually incidentally found) could have Cushing’s disease. This is a critical distinction to make as management of Cushing’s disease includes surgical resection; whereas, there may be no acute indication for surgery with a small nonfunctional adenoma.
In general, only patients with multiple and progressive clinical features of hypercortisolism (previously described in Table 2 ) should be considered for biochemical screening. Before undertaking a biochemical evaluation, a thorough history is essential as exogenous glucocorticoid use represents the most common cause of clinical hypercortisolism (“iatrogenic Cushing’s”). Endogenous hypercortisolism from Cushing’s disease is characterized by loss of appropriate feedback control leading to increased cortisol, loss of normal diurnal rhythm of cortisol secretion, and reduced ability of steroids (endogenous or exogenous) to inhibit ACTH secretion. These characteristics can be assessed using 3 biochemical tests: (1) 24-hour urinary free cortisol (UFC) excretion (assesses total cortisol production), (2) late night salivary cortisol collection (assesses whether diurnal rhythm is present), and (3) 1 mg dexamethasone suppression test (assesses whether exogenous steroid can appropriately suppress ACTH/cortisol production). In general, at least 2 tests must be unequivocally abnormal (eg, 24 h UFC >3× upper limit of normal) to establish the diagnosis of Cushing’s syndrome. Diagnosis of Cushing’s syndrome via these tests typically occurs in conjunction with an endocrinologist; a referral to an endocrinologist should be considered for any patients with a history of, or physical features suggestive of, hypercortisolism.
Conclusively establishing the diagnosis of Cushing’s disease can be a challenging endeavor and a full discussion is beyond the scope of this article. Briefly, patients with unequivocally positive screening tests should have ACTH measured (to confirm the pituitary origin of hypercortisolism) followed by MRI of the pituitary. In cases where MRI is equivocal, then several options to localize ACTH overproduction to the pituitary exist, including high-dose dexamethasone suppression tests or sampling of ACTH levels in the inferior petrosal sinus. While invasive, inferior petrosal sinus sampling has a sensitivity and specificity of greater than 90% and is the preferred option at pituitary centers where technical expertise and experience exists.
Prolactinoma
Diagnosis of prolactinoma is straightforward when prolactin is markedly increased (eg, >500 μg/L) with a visible pituitary adenoma on MRI. In general, prolactin levels parallel tumor size; most prolactinomas greater than 1 cm have prolactin concentrations greater than 250 μg/L. Hyperprolactinemia in the absence of a pituitary tumor should prompt assessment for other causes of hyperprolactinemia (listed in Table 3 ). Drug-induced hyperprolactinemia is relatively common; increases are typically less than 200 μg/L, although certain atypical antipsychotics such as risperidone can rarely cause increases to greater than 200 μg/L.



